Ramon C. Sun, Mitali Fadia, Jane E. Dahlstrom, Christopher R. Parish, Philip G. Board, Anneke C. Blackburn
R.C. Sun, P. G. Board, A. C. Blackburn
Molecular Genetics Group, John Curtin School of Medical Research, Australian National University, P.O. Box 334, Canberra 2601, Australia e-mail: [email protected]
M.Fadia, J. E. Dahlstrom
Department of Anatomical Pathology, Canberra Hospital and Australian National University Medical School, Woden ACT 2606, Australia
C.R. Parish
Cancer and Vascular Biology Group, John Curtin School of Medical Research, Australian National University, Canberra ACT 0200, Australia
Ricevuto: 17 aprile 2009
Accettato: 2 giugno 2009
Pubblicato: 19 giugno 2009
Abstract
Il fenotipo glicolitico è un fenomeno diffuso nelle forme tumorali solide, compreso il cancro al seno. Il dicloroacetato (DCA) è stato recentemente proposto come un agente antitumorale nuovo e relativamente non tossico, in grado di invertire il fenotipo glicolitico nelle cellule tumorali attraverso l’inibizione della piruvato deidrogenasi chinasi. Abbiamo esaminato l’effetto del DCA contro le cellule del cancro al seno, anche in un modello in vivo altamente metastatico. La crescita di diverse linee cellulari di cancro al seno è stata inibita dal DCA in vitro. Da un ulteriore esame delle cellule di adenocarcinoma mammario di ratto 13762 MAT è emerso che l’inversione del fenotipo glicolitico da parte del DCA è correlata all’inibizione della proliferazione senza alcun aumento della morte cellulare. Questo nonostante un piccolo ma significativo aumento dell’attività della caspasi 3/7, che potrebbe sensibilizzare le cellule tumorali ad altri fattori apoptotici. In vivo, il DCA ha causato una riduzione del 58% del numero di metastasi polmonari osservate macroscopicamente dopo l’iniezione di 13762 cellule MAT nella vena caudale dei ratti (P = 0,0001, n ≥ 9 per gruppo). Questi risultati dimostrano che il DCA ha proprietà antiproliferative oltre a proprietà pro-apoptotiche e può essere efficace contro la malattia altamente metastatica in vivo, evidenziando il suo potenziale per l’uso clinico.
Parole chiave: Dicloroacetato, Cancro al seno, Glicolisi, Metastasi, Modello animale
© Springer Science+Business Media, LLC. 2009
INTRODUZIONE
Il fenotipo glicolitico, spesso indicato come effetto Warburg, è un fenomeno diffuso nella maggior parte delle forme tumorali in cui si verificano alti tassi di assorbimento di glucosio e glicolisi, mentre la respirazione mitocondriale è repressa, nonostante la presenza di ossigeno. Si ritiene che questa caratteristica metabolica sia acquisita per la produzione di ATP durante l’evoluzione anaerobica del tumore; tuttavia, le prove indicano sempre più che il fenotipo glicolitico è accompagnato da cambiamenti dell’espressione genica che sono intimamente legati ai processi tumorigenici, come la resistenza all’apoptosi e l’aumento del potenziale metastatico [1,2].
L’esistenza del fenotipo glicolitico nel cancro al seno è stata ben descritta. Un indice bioenergetico cellulare (BEC) alterato e un profondo spostamento verso un fenotipo glicolitico potenziato sono stati riportati nei tumori al seno rispetto alle biopsie di tessuto mammario normale accoppiate e correlati alla sopravvivenza globale e libera da malattia delle pazienti [3,4]. L’invasività di diverse linee cellulari di carcinoma mammario è stata correlata a un livello costitutivo più elevato del fattore di trascrizione HIF-1α in condizioni di normossia e a una minore induzione di HIF-1α in ipossia, nonché a una maggiore produzione di lattato [5]. Dal punto di vista immunoistochimico, nei focolai microinvasivi di carcinoma duttale in situ (DCIS) è stata osservata una upregulation di due marcatori del fenotipo glicolitico (il trasportatore di glucosio GLUT1 e lo scambiatore Na+/H+ NHE-1), indicando che l’adattamento all’ipossia e all’acidosi può rappresentare eventi chiave nella transizione dal carcinoma mammario in situ a quello invasivo [6]. Pertanto, l’inversione del fenotipo glicolitico per la prevenzione delle metastasi e delle recidive del carcinoma mammario è una strategia di trattamento rilevante.
La piruvato deidrogenasi (PDH) regola la conversione del piruvato in acetil Co-A e può quindi controllare il flusso di metaboliti dalla glicolisi al ciclo dell’acido citrico e quindi la generazione di ATP da parte dei mitocondri. La PDH è regolata dalla piruvato deidrogenasi chinasi (PDK) che fosforila e inattiva la PDH [7]. Il dicloroacetato (DCA) inibisce la PDK ed è stato recentemente proposto come agente antitumorale nuovo e relativamente non tossico [8]. È stato dimostrato che il DCA inverte il fenotipo glicolitico in diverse linee cellulari tumorali, depolarizzando il potenziale di membrana mitocondriale interno iperpolarizzato a livelli normali e aumentando il metabolismo mitocondriale [8,9]. Poiché il DCA agisce su un cambiamento in atto durante la tumorigenesi, può essere efficace contro le cellule tumorali senza tossicità per le cellule normali. Il DCA è attualmente in fase III di sperimentazione clinica per il trattamento dell’acidosi lattica cronica nei disordini mitocondriali congeniti [10,11] e ha quindi il potenziale per entrare rapidamente in clinica per altre applicazioni, avendo superato i test di tossicità di fase I/II nell’uomo [12]. Sono in corso studi clinici che valutano la sua tossicità nei pazienti oncologici (http://www.clinicaltrials.gov); tuttavia, sono necessari esperimenti controllati per comprendere le attività antitumorali del DCA, al fine di determinare quali tumori e quali pazienti siano più adatti a essere trattati con il DCA.
In questo studio è stata utilizzata una linea cellulare di adenocarcinoma mammario di ratto per esaminare l’effetto del DCA sia in vitro che in vivo. I risultati suggeriscono un meccanismo d’azione del DCA in queste cellule come agente antiproliferativo piuttosto che come agente che induce l’apoptosi e dimostrano che il DCA può essere efficace in vivo nel ridurre la crescita delle cellule tumorali metastatiche, aumentando la sua importanza nel trattamento del cancro al seno.
Materiali e metodi
Coltura cellulare
13762 Le cellule di adenocarcinoma mammario di ratto MAT (cellule MAT) sono state mantenute in vitro come precedentemente descritto [13]. Le cellule V14 sono derivate da un adenocarcinoma mammario spontaneo insorto in un topo BALB/c-Trp53+/- [14].
Crescita cellulare
Le cellule sono state esposte a 1-5 mM di DCA (Sigma Chemical Co St. Louis, MO) per 1-4 giorni in piastre a 96 pozzetti, con rinnovo giornaliero del terreno e del DCA, e la vitalità cellulare è stata misurata mediante captazione di rosso neutro [15].
Apoptosi
Le attività della caspasi 3/7 nelle cellule MAT sono state valutate utilizzando il saggio Caspase-Glo 3/7 (Promega Corp., Madison, WI) secondo le istruzioni del produttore. L’apoptosi è stata quantificata mediante citometria a flusso dopo aver colorato le cellule con Annexina-V marcata con FITC (BD Pharmingen, NJ) e ioduro di propidio (PI) (Sigma Chemical Co St. Louis, MO).
Proliferazione
Le cellule sono state colorate con 5 μM di carbossifluoresceina succinimidil estere (CFSE) ed esaminate mediante analisi di ordinamento cellulare attivato dalla fluorescenza [16].
Metabolismo cellulare
I livelli interni di ATP nelle cellule MAT sono stati valutati utilizzando il saggio CellTiter-Glo (Promega Corp., Madison, WI) secondo le istruzioni del produttore. I livelli di lattato extracellulare sono stati determinati spettrofotometricamente misurando la conversione di NAD in NADH a 340 nm da parte della lattato deidrogenasi in estratti di terreno neutralizzati con acido perclorico [17].
13762 Metastasi di cellule di MAT in vivo
Gli esperimenti sugli animali sono stati condotti con l’approvazione del Comitato per la sperimentazione etica sugli animali dell’Australian National University, secondo le linee guida stabilite dall’Australian National Health and Medical Research Committee. A tre gruppi di ratti femmina Fischer 344 (10-13 settimane di età) sono state iniettate 2 ×105 cellule MAT 13762 nella vena laterale della coda [13]. Il gruppo 1 (controllo) non è stato trattato. Prima dell’iniezione, i gruppi 2 (dose bassa) e 3 (dose alta) hanno ricevuto DCA somministrato per via orale in acqua potabile a 0,2 g/l (23 mg/kg) per 7 giorni per esaurire l’attività di GSTZ1 e massimizzare la biodisponibilità di DCA [18]. Il giorno dell’iniezione delle cellule la dose orale è stata aumentata a 0,75 g/l (consumo medio di acqua 115 ml/kg/die), corrispondente a una dose giornaliera di 86 mg/kg senza alterare significativamente il consumo di acqua (controllo 120 ml/kg/die). I ratti del gruppo 3 sono stati sottoposti a un ulteriore trattamento con DCA (dose elevata), ricevendo 200 mg/kg/die per via intraperitoneale (i.p.) in soluzione fisiologica tamponata con fosfato (neutralizzata e sterilizzata in filtro), con la prima iniezione somministrata circa 2 ore prima dell’iniezione delle cellule. Estrapolando dai dati pubblicati [18,19], si stima che il dosaggio orale determini concentrazioni plasmatiche dell’ordine di 0,5-1 mM di DCA, mentre si stima che l’ulteriore somministrazione di 200 mg/kg per via i.p. aumenti di circa tre volte fino a 1,5-3,0 mM.
I ratti sono stati uccisi 14 giorni dopo l’iniezione delle cellule tumorali e i polmoni sono stati fissati nella soluzione di Bouins. Il numero di metastasi polmonari è stato valutato al microscopio a dissezione. I due lobi più grandi di ciascun polmone sono stati successivamente inclusi in paraffina e colorati per la valutazione microscopica. Sono stati valutati il numero di lesioni microscopiche, le loro dimensioni e il numero di mitosi per campo ad alta potenza (hpf). È stata riportata la presenza o l’assenza di necrosi tumorale ed è stata registrata la densità dei linfociti associati al tumore (occasionale/moderata/grave).
Attività di GSTZ
Il DCAviene metabolizzato a gliossilato dalla glutatione transferasi GSTZ1-1 nel fegato, ma il DCA può inibire il proprio catabolismo formando un complesso enzima-substrato inattivo con GSTZ1 [20]. Per garantire un’efficace somministrazione di DCA, l’attività di GSTZ1 è stata misurata nel fegato di ratto secondo il metodo precedentemente descritto [21].
Analisi statistica
I dati FACS sono stati acquisiti con il pacchetto software Cell Quest (BD Bioscience, Rockville, MD) e analizzati con FlowJo (Tree Star Inc, OR). I calcoli sono stati eseguiti con il pacchetto software GraphPad Prism® e il t-test di Student è stato applicato per valutare le differenze tra i gruppi trattati con DCA e quelli di controllo. Un valore P inferiore a 0,05 è stato considerato statisticamente significativo. I dati sono rappresentati come media ± deviazione standard.
Risultati
IlDCA inibisce la crescita delle cellule di cancro al seno
Per studiare la sensibilità delle cellule di cancro al seno al DCA, abbiamo trattato un gruppo di linee cellulari di cancro al seno con 5 mM di DCA (Fig. 1a). Le cellule MCF-7, T-47D, 13762 MAT e V14 hanno mostrato una riduzione del 60-80% del numero di cellule al quarto giorno di trattamento, mentre le cellule 4T1 sono risultate insensibili. Al contrario, il DCA non ha avuto alcun effetto sulla crescita di una linea cellulare di controllo non cancerosa, la MCF-10A.
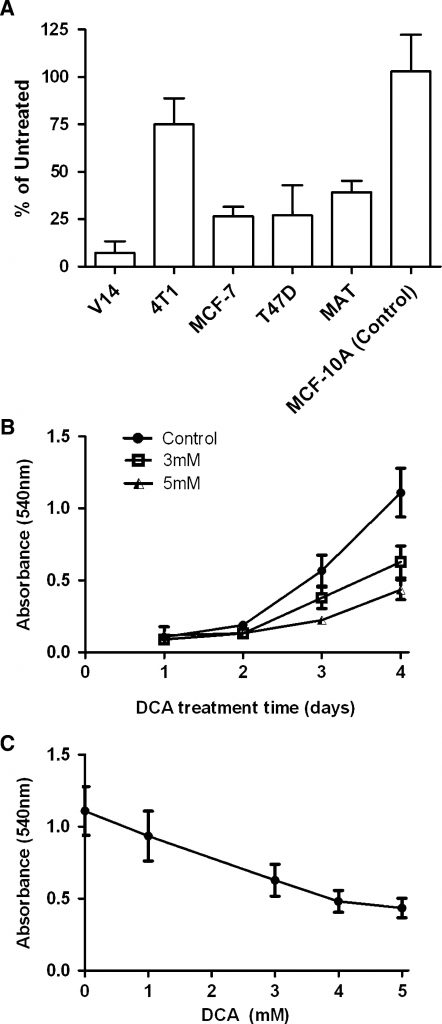
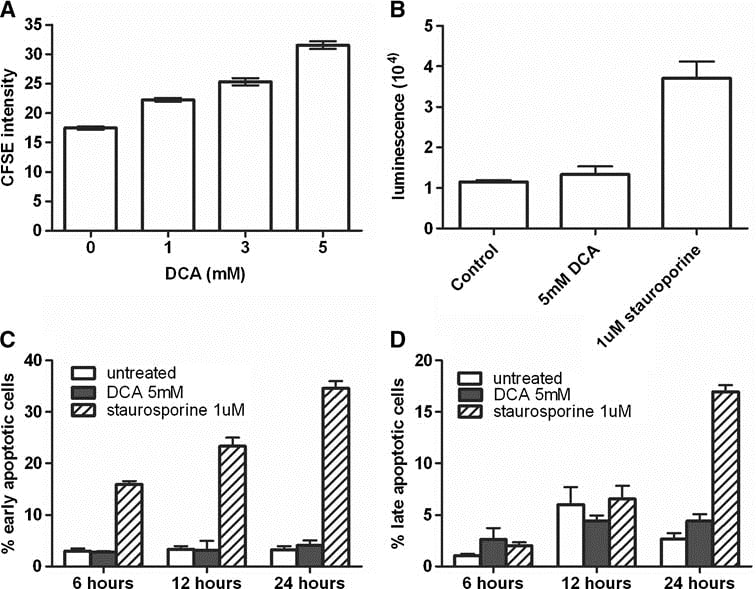
L’effetto del DCA sulle cellule MAT è stato ulteriormente esaminato sia in vitro che in vivo. La risposta delle cellule MAT al DCA è stata dipendente dal tempo e dalla dose (Fig. 1b, c). L’effetto più forte è stato osservato il giorno 4, quando le cellule MAT trattate con 5 mM di DCA avevano il 68 ± 5% di cellule in meno rispetto alle colture di controllo (n = 3, P < 0,0001). Per determinare la ragione della diminuzione del numero di cellule, sono state misurate la proliferazione cellulare e l’apoptosi. Utilizzando il saggio di proliferazione cellulare CFSE, è emerso che dopo 3 giorni le cellule MAT trattate con 5 mM di DCA mostravano una fluorescenza significativamente più alta rispetto alle cellule non trattate (P = 0,0009; n = 3), indicando una riduzione della divisione cellulare (Fig. 2a). Ciò era evidente anche a 1 mM di DCA. Al contrario, l’apoptosi non è stata aumentata dal DCA (Fig. 2b-d). Il trattamento con 5 mM di DCA ha mostrato un piccolo (15%) ma statisticamente significativo aumento dell’attività della caspasi 3/7 dopo 3 ore (Fig. 2b); tuttavia, si tratta di un aumento minimo rispetto al controllo positivo con staurosporina (2,2 volte). Anche la colorazione con Annexin V e PI ha indicato che il trattamento con DCA 5 mM non è riuscito a indurre l’apoptosi nelle cellule MAT anche dopo 24 ore di incubazione (Fig. 2c, d).
IlDCA inverte il fenotipo glicolitico
Il trattamento delle cellule MAT con 5 mM di DCA per 30 minuti ha determinato un aumento del 18 ± 3% dei livelli di ATP totale (n = 3, P = 0,009), e questo effetto è persistito a 3 ore. Dopo 12 ore di trattamento con DCA, la concentrazione extracellulare di lattato è diminuita del 16,3 ± 5,3% (n = 4, P = 0,01). Questi dati confermano l’inversione del fenotipo glicolitico delle cellule MAT da parte del DCA.
IlDCA ha ridotto la crescita tumorale in vivo
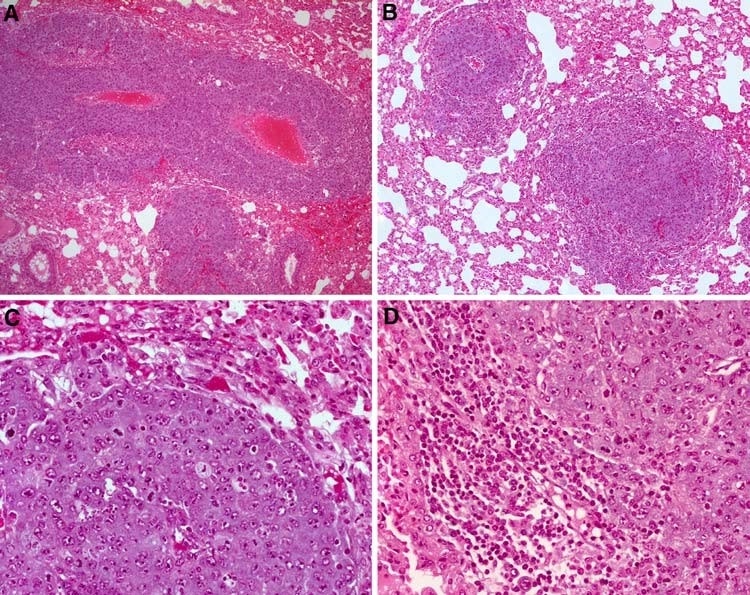
Dopo l’iniezione i.v. di cellule MAT, non c’è stata alcuna variazione nel numero di metastasi nei ratti del gruppo a bassa dose di DCA (che hanno ricevuto ∼86 mg/kg di DCA solo per via orale). Al contrario, i ratti del gruppo DCA ad alta dose (che ricevevano iniezioni giornaliere i.p. di 200 mg/kg/die di DCA in aggiunta al DCA orale) hanno mostrato una riduzione significativa del 58 ± 17% del numero di tumori polmonari osservati macroscopicamente (P = 0,0001, n ≥ 9 per gruppo) (Fig. 3). A livello microscopico, tuttavia, il numero di lesioni è rimasto invariato nei tre gruppi (6,4 ± 2,8, 7,1 ± 3,4 e 6,2 ± 3,2 per 5 campi ad alta potenza per controllo, bassa e alta dose, rispettivamente). Le lesioni nei ratti trattati con DCA ad alta dose hanno sviluppato meno necrosi tumorale e hanno avuto una conta mitotica più alta (9,4 ± 7,0 vs 20,2 ± 9,2 per 5 hpf nel controllo e nell’alta dose di DCA, rispettivamente, P = 0,03) (Fig. 4). I corpi apoptotici non erano presenti in nessuno dei gruppi. Il trattamento con DCA ha portato anche a una moderata infiltrazione linfocitaria soprattutto ai margini dei tumori, mentre nel gruppo di controllo erano presenti solo occasionali linfociti associati al tumore (Fig. 4).
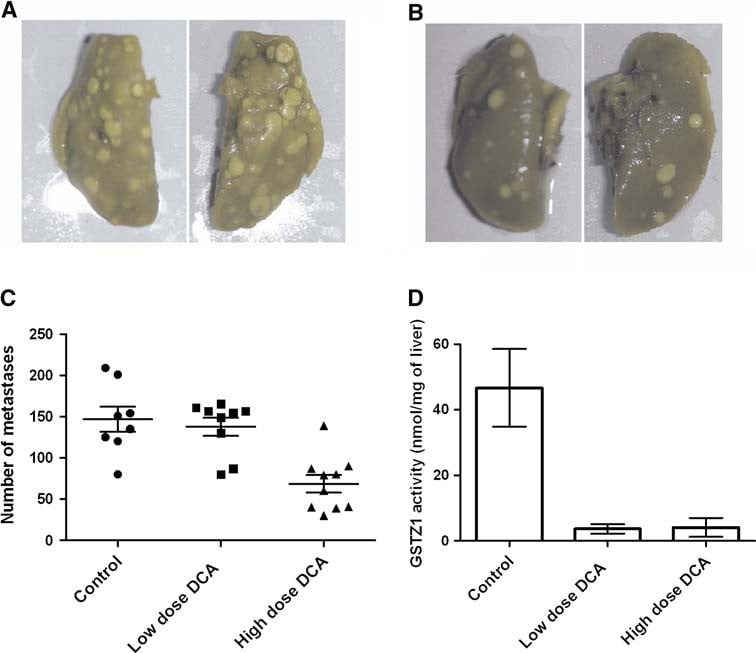
L’attività di GSTZ1-1 è stata misurata nel fegato di ratti trattati con DCA e portatori di tumore per confermare l’esposizione dei ratti al DCA. I gruppi di trattamento a bassa e alta dose hanno mostrato una diminuzione del 93% e del 95%, rispettivamente, dell’attività GSTZ1-1 nel fegato dopo il trattamento con DCA, indicando una rimozione quasi completa dell’attività GSTZ1-1 a entrambe le dosi.
Discussione
Il fenotipo glicolitico è quasi universalmente presente nei carcinomi mammari ed è associato a focolai microinvasivi e a una peggiore sopravvivenza [2-5]. Il DCA è un inibitore della PDK ed è in grado di invertire il fenotipo glicolitico. In questo studio, riportiamo che diverse linee cellulari di carcinoma mammario sono sensibili al DCA, con inibizione della crescita osservata per diversi giorni di trattamento (Fig. 1). Gli studi in vitro sulle cellule MAT hanno dimostrato chiaramente che ciò è dovuto all’inibizione della proliferazione, senza segni di apoptosi o morte cellulare (Fig. 2). Ciò contrasta con gli studi pubblicati finora sul trattamento con DCA di cellule di cancro dell’endometrio, della prostata e del polmone, dove nella maggior parte dei casi è stato riportato un aumento dell’apoptosi senza effetti sulla distribuzione del ciclo cellulare [9,22], o un aumento dell’apoptosi accompagnato da una diminuzione della proliferazione [8]. Solo due delle sei linee cellulari riportate hanno mostrato un cambiamento nella distribuzione del ciclo cellulare dopo il trattamento con DCA, una con un arresto G0/G1 e l’altra con un arresto S e uno G2/M [9,22]. Sebbene il DCA inibisca la crescita cellulare in un’ampia gamma di cellule tumorali, il meccanismo sembra dipendere dalla linea cellulare. È stata osservata anche una mancanza di apoptosi nella linea cellulare di cancro al seno umano T-47D dopo il trattamento con DCA (dati non mostrati), indicando che questa risposta non è esclusiva delle cellule MAT, ma potrebbe essere una caratteristica delle cellule di cancro al seno. Si tratta di una possibilità intrigante che sarà oggetto di ulteriori indagini. In alternativa, la mancanza di apoptosi potrebbe essere dovuta ad alti livelli di proteine di sopravvivenza cellulare, come Bcl-2, survivin e PUMA, nelle particolari linee cellulari analizzate. L’espressione di questi fattori di sopravvivenza è stata ridotta dal trattamento con DCA in cellule di cancro della prostata, del polmone e dell’endometrio [8,22] e ciò potrebbe contribuire alla risposta apoptotica osservata.
La sensibilità delle linee cellulari di cancro al seno al DCA variava dal 20 all’80% di inibizione della crescita cellulare in 4 giorni di trattamento con 5 mM (Fig. 1), con le cellule 4T1 meno sensibili. La sensibilità al DCA può essere determinata da diversi fattori, tra cui la capacità di metabolizzare il DCA tramite GSTZ1 o la sovraespressione di diverse isoforme PDK. Esistono quattro isoforme di PDK, con Kis per il DCA rispettivamente di 1,0, 0,2, 8,0 e 0,5 mM [23]. Mentre le PDK1, 2 e 4 sarebbero inibite dalle concentrazioni di DCA utilizzate in questo studio, la PDK3 non lo sarebbe. L’espressione di PDK3 è normalmente limitata ai testicoli [23], anche se l’espressione e l’induzione di PDK3 da parte dell’ipossia sono state riportate per diverse linee cellulari tumorali [24]. Sono in corso studi per correlare l’espressione di PDK con la sensibilità al DCA e per determinare quali tumori sono più efficacemente colpiti dal DCA.
È stato osservato anche un piccolo aumento delle attività della caspasi 3/7. Questo potrebbe essere dovuto alla reattività della caspasi. Questo potrebbe essere dovuto alla riattivazione della catena di trasporto degli elettroni da parte del DCA e all’aumento della produzione di specie reattive dell’ossigeno e dell’azoto nei mitocondri [25]. Questo aumento del livello basale dell’attività delle caspasi potrebbe non essere sufficiente per indurre l’apoptosi; tuttavia, potrebbe indicare che il DCA potrebbe essere utilizzato per sensibilizzare le cellule tumorali verso altri fattori scatenanti l’apoptosi, come l’ipossia, le radiazioni o altri agenti chemioterapici. Queste potenziali sinergie richiedono ulteriori analisi.
In vivo, l’inversione del fenotipo glicolitico attraverso l’accoppiamento piruvato-acetil CoA della glicolisi e della respirazione mitocondriale è stata precedentemente dimostrata efficace contro la crescita del tumore primario in due modelli [1,8]. Abbiamo dimostrato l’efficacia del DCA contro la malattia metastatica in vivo nel modello cellulare MAT. Mentre il numero macroscopico di lesioni polmonari è stato ridotto dal DCA ad alte dosi, il numero di lesioni microscopiche non è stato modificato, suggerendo che l’effetto principale del DCA è stato sulle dimensioni dei tumori, piuttosto che una riduzione del numero di cellule in grado di stabilirsi come tumori nei polmoni. Anche l’osservazione di una minore incidenza di necrosi nei tumori trattati con DCA è coerente con un meccanismo di inibizione della crescita, come osservato in vitro. L’aumento del numero di mitosi sembra inizialmente in contrasto con questo dato, suggerendo tassi di proliferazione più elevati; tuttavia, suggeriamo che ciò possa essere dovuto all’arresto del ciclo cellulare da parte del DCA prima dell’anafase, che porta a un accumulo di cellule presenti come figure mitotiche. È interessante notare che negli animali trattati con alte dosi di DCA si è registrato un aumento dei linfociti associati al tumore. Una risposta immunitaria più forte contro i tumori potrebbe essere promossa dalla riduzione dei livelli di lattato tumorale ottenuta con il trattamento con DCA, poiché è stato dimostrato che elevate concentrazioni di acido lattico riducono la funzione delle cellule T [26]. Gli esperimenti con le cellule V14 in vivo suggeriscono che le cellule V14 sono sensibili al DCA in modo simile alle cellule MAT, con una riduzione della crescita del tumore primario e un aumento della presenza di linfociti (dati non mostrati), indicando che questi effetti in vivo non sono esclusivi delle cellule MAT.
L’uso di agenti a concentrazioni millimolari è spesso considerato insostenibile. Tuttavia, livelli sierici millimolari di DCA (0,3-1 mM) sono stati mantenuti cronicamente nei pazienti mediante somministrazione orale di DCA a 25 mg/kg/die [27]. Nel trattamento acuto dei pazienti, sono stati tollerati fino a 80 mg/kg i.v. durante il trapianto di fegato [28]. Sebbene la dose efficace di DCA in vivo in questo modello di ratto fosse elevata, la concentrazione plasmatica stimata raggiunta in vivo (1,5-3 mM, vedi Metodi) è simile a quella degli esseri umani che ricevono 25 mg/kg/die, ed è quindi rilevante per il contesto clinico. Questo intervallo di concentrazioni plasmatiche si correla anche con l’inibizione della proliferazione in vitro (a partire da 1 mM, Fig. 2a) e con il Ki per l’inibizione delle PDK da parte del DCA [23], a sostegno della proposta che la PDK sia il bersaglio responsabile delle attività antitumorali del DCA.
Sebbene il trattamento cronico con DCA nei pazienti con MELAS abbia provocato una certa neurotossicità periferica reversibile [10], la tossicità del DCA per i tessuti vulnerabili agli agenti citotossici classici è minima [8,29], il che lo rende un buon agente candidato per la terapia di combinazione. Ad esempio, mentre il DCA inibisce irreversibilmente la GSTZ1 (Fig. 2d), i topi trattati con DCA non hanno mostrato la deplezione dei linfociti osservata nei topi geneticamente carenti di GSTZ [21,30]. Pertanto, l’inversione del fenotipo glicolitico nel carcinoma mammario attraverso l’inibizione della PDK con il DCA è una strategia antitumorale promettente e dimostra anche una potenziale applicazione per gli inibitori alternativi della PDK attualmente in fase di sviluppo [31]. Tuttavia, sono necessari ulteriori studi meccanici per comprendere la relazione causale tra il fenotipo glicolitico e le caratteristiche del tumore, al fine di poter indirizzare in modo efficace il metabolismo delle cellule tumorali a fini terapeutici.
Ringraziamenti
Questa ricerca è stata sostenuta da una sovvenzione della National Breast Cancer Foundation Australia e dal NHMRC 366787 R.D. Wright Career Development Award.
RIFERIMENTI
1 Fantin VR, St-Pierre J, Leder P (2006) L’attenuazione dell’espressione di LDH-a scopre un legame tra glicolisi, fisiologia mitocondriale e mantenimento del tumore. Cancer Cell 9:425-4342 Gatenby R, Gillies RJ (2007) Glycolysis in cancer: a potential target for therapy. Int J Biochem Cell Biol 39:1358-1366
3 Isidoro A, Martnez M, Fernndez PL, Ortega AD, Santamara G, Chamorro M, Reed JC, Cuezva JM (2004) Alteration of the bioenergetic phenotype of mitochondria is a hallmark of breast, gastric, lung and oesophageal cancer. Biochem J 378:17-20
4 Isidoro A, Casado E, Redondo A, Acebo P, Espinosa E, Alonso AM, Cejas P, Hardisson D, Fresno Vara JA, Belda-Iniesta C, Gonzlez-Barn M, Cuezva JM (2005) I carcinomi mammari soddisfano l’ipotesi di Warburg e forniscono marcatori metabolici della prognosi del cancro. Carcinogenesi 26:2095-2104
5 Robey IF, Lien AD, Welsh SJ, Baggett BK, Gillies RJ (2005) Hypoxia-inducible factor-1α and the glycolytic phenotype in tumors. Neoplasia 7:324-330
6 Gatenby RA, Smallbone K, Maini PK, Rose F, Averill J, Nagle RB, Worrall L, Gillies RJ (2007) Cellular adaptations to hypoxia and acidosis during somatic evolution of breast cancer. Br J Cancer 97:646-653
7 Gudi R, Bowker-Kinley MM, Kedishvili NY, Zhao Y, Popov KM (1995) Diversity of the pyruvate dehydrogenase kinase gene family in humans. J Biol Chem 270:28989-28994
8 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED (2007) A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11:37-51
9 Wong JY, Huggins GS, Debidda M, Munshi NC, Vivo ID (2008) Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol Oncol 109:394-402
10 Kaufmann P, Engelstad K, Wei Y, Jhung S, Sano MC, Shungu DC, Millar WS, Hong X, Gooch CL, Mao X, Pascual JM, Hirano M, Stacpoole PW, DiMauro S, Vivo DCD (2006) Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurologia 66:324-330
11 Stacpoole PW, Kerr DS, Barnes C, Bunch ST, Carney PR, Fennell EM, Felitsyn NM, Gilmore RL, Greer M, Henderson GN, Hutson AD, Neiberger RE, O’Brien RG, Perkins LA, Quisling RG, Shroads AL, Shuster JJ, Silverstein JH, Theriaque DW, Valenstein E (2006) Studio clinico controllato del dicloroacetato per il trattamento dell’acidosi lattica congenita nei bambini. Pediatria 117:1519-1531
12 Pearson H (2007) I pazienti affetti da cancro scelgono un farmaco non approvato. Nature 446:474-475
13 Parish CR, Freeman C, Brown KJ, Francis DJ, Cowden WB (1999) Identificazione di inibitori della crescita tumorale e delle metastasi a base di oligosaccaridi solfatati utilizzando nuovi saggi in vitro per l’angiogenesi e l’attività dell’eparanasi. Cancer Res 59:3433-3441
14 Blackburn AC, McLary SC, Naeem R, Luszcz J, Stockton DW, Donehower LA, Mohammed M, Mailhes JB, Soferr T, Naber SP, Otis CN, Jerry DJ (2004) La perdita di eterozigosi avviene tramite ricombinazione mitotica nei topi Trp53+/- e si associa alla suscettibilità al tumore mammario del ceppo BALB/c. Cancer Res 64:5140-5147
15 Schmuck E, Cappello J, Coggan M, Brew J, Cavanaugh JA, Blackburn AC, Baker RT, Eyre HJ, Sutherland GR, Board PG (2008) La delezione di Glu155 causa una carenza di glutatione transferasi Omega 1-1 ma non altera la sensibilità al triossido di arsenico e ad altri farmaci citotossici. Int J Biochem Cell Biol 40:2553-2559
16 Quah BJ, Warren HS, Parish CR (2007) Monitoring lymphocyte proliferation in vitro and in vivo with the intracellular fluorescent dye carboxyfluorescein diacetate succinimidyl ester. Nat Protoc 2:2049-2056
17 Beutler E (1975) Red cell metabolism: a manual of biochemical methods, 2nd edn. Grune & Stratton Inc., New York
18 Saghir SA, Schultz IR (2002) Low-dose pharmacokinetics and oral bioavailability of dichloroacetate in naive and GST-zeta-depleted rats. Environ Health Perspect 110:757-763
19 Gonzalez-Leon A, Schultz IR, Xu G, Bull RJ (1997) Farmacocinetica e metabolismo del dicloroacetato nel ratto F344 dopo una precedente somministrazione in acqua potabile. Tossicologia Applicata Farmacologia 146:189-195
20 Board PG, Anders MW (2005) La glutatione transferasi zeta umana. Metodi Enzimologici 401:61-77
21 Lim CEL, Matthaei KI, Blackburn AC, Davis RP, Dahlstrom JE, Koina ME, Anders MW, Board PG (2004) I topi carenti di glutatione transferasi zeta/maleilacetoacetato isomerasi presentano una serie di alterazioni patologiche e un’elevata espressione delle glutatione transferasi di classe alfa, mu e pi. Am J Pathol 165:679-693
22 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ (2008) Il dicloroacetato (DCA) sensibilizza alle radiazioni in vitro sia le cellule di cancro alla prostata wild-type che quelle che esprimono Bcl-2. Prostata 68:1223-1231
23 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM (1998) Prove dell’esistenza di una regolazione tessuto-specifica del complesso della piruvato deidrogenasi nei mammiferi. Biochem J 329:191-196
24 Lu CW, Lin SC, Chen KF, Lai YY, Tsai SJ (2008) Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J Biol Chem 283:28106-28114
25 DiPietrantonio AM, Hsieh T, Wu JM (1999) Attivazione della caspasi 3 in cellule HL-60 esposte al perossido di idrogeno. Biochem Biophys Res Commun 255:477-482
26 Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G, Hoves S, Renner K, Timischl B, Mackensen A, Kunz-Schughart L, Andreesen R, Krause SW, Kreutz M (2007) Inhibitory effect of tumor cell-derived lactic acid on human T cells. Sangue 109:3812-3819
26 Mori M, Yamagata T, Goto T, Saito S, Momoi MY (2004) Trattamento con dicloroacetato per la citopatia mitocondriale: effetti a lungo termine nel MELAS. Brain Dev 26:453-458
28 Shangraw RE, Lohan-Mannion D, Hayes A, Moriarty RM, Fu R, Robinson ST (2008) Dichloroacetate stabilizes the intraoperative acid-base balance during liver transplantation. Trapianto di fegato 14:989-998
29StacpoolePW, Gilbert LR, Neiberger RE, Carney PR, Valenstein E, Theriaque DW, Shuster JJ (2008) Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. Pediatria 121:e1223-e1228
30 Theodoratos A, Tu WJ, Cappello J, Blackburn AC, Matthaei K, Board PG (2009) Phenylalanine-induced leucopenia in genetic and dichloroacetic acid generated deficiency of glutathione transferase zeta. Biochimica Farmacologia 77:1358-1363
31 Mayers RM, Leighton B, Kilgour E (2005) Inibitori della PDH chinasi: una nuova terapia per il diabete di tipo II? Biochem Soc Trans 33:367-370
Contenuti correlati: