Cecilie Abildgaard1, Christina Dahl1, Ahmad Abdul-Al1, Annette Christensen1 e Per Guldberg1
1 Centro di ricerca della Società danese per il cancro, Copenaghen, Danimarca
Corrispondenza: Per Guldberg, email: [email protected]
Ricevuto: 19 aprile 2017
Accettato: 19 luglio 2017
Pubblicato: 24 agosto 2017
Abstract
La disregolazione del metabolismo durante la progressione del melanoma è strettamente associata all’acquisizione di alterazioni genetiche ed epigenetiche nei regolatori delle vie metaboliche. Il recettore beta dell’acido retinoico (RARβ) è epigeneticamente silenziato in un’ampia percentuale di melanomi, ma non è stato stabilito un legame tra RARβ e il ricablaggio metabolico del melanoma. Qui dimostriamo che nei melanociti umani primari, l’acido retinoico all-trans (un agonista di RARβ) induce un’inibizione della crescita accompagnata da una diminuzione del metabolismo glicolitico e ossidativo, mentre l’inibizione selettiva di RARβ porta a un aumento del tasso glicolitico basale e a una maggiore sensibilità all’inibizione della glicolisi. Nelle cellule di melanoma, l’inibizione di RARβ ha promosso una minore respirazione mitocondriale e una maggiore attività glicolitica, con conseguente stress energetico e attivazione del sensore energetico AMP-activated protein kinase. Questo spostamento metabolico ha aumentato la sensibilità sia all’inibizione glicolitica sia alla stimolazione del metabolismo mitocondriale con dicloroacetato, un inibitore della piruvato deidrogenasi chinasi. Nelle cellule di melanoma portatrici della mutazione BRAFV600E, l’attivazione di RARβ ha antagonizzato l’effetto dell’inibitore di BRAF PLX4032 (vemurafenib). Nel complesso, questi dati suggeriscono che la segnalazione di RARβ è coinvolta nella regolazione del metabolismo cellulare nel melanoma e può rappresentare un potenziale bersaglio nelle strategie di trattamento combinato.
Parole chiave: melanoma, metabolismo del cancro, recettore dell’acido retinoico β, respirazione mitocondriale, dicloroacetato
Abbreviazioni: ATRA: acido retinoico all-trans; DCA: dicloroacetato; ECAR: tasso di acidificazione extracellulare; OCR: tasso di consumo di ossigeno; ROS: specie reattive dell’ossigeno
© Abildgaard et al. Questo è un articolo ad accesso libero distribuito secondo i termini della Creative Commons Attribution License 3.0 (CC BY 3.0), che ne consente l’uso, la distribuzione e la riproduzione illimitati su qualsiasi supporto, a condizione che vengano citati l’autore e la fonte originale.
INTRODUZIONE
Il melanoma, la forma più letale di cancro della pelle, causa ogni anno 50.000 decessi e l’incidenza continua ad aumentare in tutto il mondo. Mentre il melanoma cutaneo primario è curabile con la chirurgia, la forma più avanzata della malattia (stadio IV) è associata a una sopravvivenza a 10 anni del 10-15% [1], il che riflette la sua nota resistenza alla terapia antitumorale convenzionale. I recenti progressi terapeutici includono gli inibitori del checkpoint immunitario e le terapie che hanno come bersaglio gli oncogeni o gli effettori a valle della via MAPK (ad esempio, gli inibitori di BRAF e MEK). Tuttavia, lo sviluppo di una resistenza acquisita ai farmaci porta alla ricaduta nella maggior parte dei casi [2, 3].
Il melanoma si sviluppa da cellule produttrici di melanina, chiamate melanociti, attraverso l’acquisizione di molteplici alterazioni genomiche. I driver più comuni del melanoma includono mutazioni attivanti in BRAF e NRAS e mutazioni inattivanti o delezioni in CDKN2A (che codifica p16INK4A e p14ARF), PTEN e TP53 [4]. Recenti evidenze suggeriscono che una funzione comune ad alcuni di questi geni è il controllo del metabolismo cellulare [5, 6]. Durante la progressione del melanoma, il metabolismo cellulare viene riprogrammato, il che implica uno spostamento dalla respirazione mitocondriale alla glicolisi aerobica, con conseguente aumento del consumo di glucosio e della produzione di acido lattico (effetto Warburg) [7]. Diversi rapporti basati su modelli in vitro e in vivo di melanoma e studi clinici su pazienti affetti da melanoma hanno dimostrato un legame tra mutazioni attivanti il codone V600 di BRAF (più comunemente BRAFV600E) e la glicolisi aerobica [8-10]. A livello molecolare, BRAFV600E regola la fosforilazione ossidativa sopprimendo il regolatore principale della biogenesi mitocondriale, PGC1α, attraverso l’inibizione del fattore di trascrizione associato alla microftalmia (MITF). Al contrario, l’inibizione di BRAFV600E porta a una dipendenza ossidativa attraverso l’induzione di PGC1α e l’aumento della respirazione mitocondriale [11]. La corrispondente diminuzione dell’attività glicolitica può essere visualizzata mediante scansione PET-CT nei pazienti con melanoma trattati con inibitori di BRAF, mostrando una ridotta captazione di glucosio nel tessuto tumorale [10]. Gli studi clinici di fase III dell’inibitore di BRAFV600E vemurafenib (PLX4032) hanno dimostrato un miglioramento della sopravvivenza globale e libera da progressione nei pazienti con melanoma metastatico [12]. Gli inibitori mitocondriali sono stati proposti come utili coadiuvanti degli inibitori della via BRAF per migliorare l’effetto o prevenire lo sviluppo della resistenza ai farmaci [13-15].
Oltre ai fattori genetici ben caratterizzati, il genoma del melanoma contiene numerose alterazioni epigenetiche. Uno dei bersagli epigenetici ricorrenti nel melanoma è RARB che codifica il recettore beta dell’acido retinoico (RARβ), silenziato dall’ipermetilazione del promotore nel 45-70% dei melanomi cutanei [16, 17]. Nelle cellule del lignaggio melanocitico, RARβ media l’inibizione della crescita e della melanogenesi indotta dall’acido retinoico (vitamina A), un marcatore della differenziazione melanocitica [18]. In precedenza abbiamo dimostrato che l’attivazione di RARβ nei melanociti induce l’upregolazione di p14ARF [17], che protegge dalla disfunzione mitocondriale e dallo stress ossidativo [19]. Qui dimostriamo che i melanociti umani rispondono all’attivazione di RARβ riducendo il metabolismo ossidativo, potenzialmente come parte di una risposta di differenziazione. Nelle cellule di melanoma, l’attivazione di RARβ antagonizza l’effetto di PLX4032, mentre l’inibizione di RARβ induce dipendenza glicolitica e stress energetico, rendendo le cellule vulnerabili al trattamento con l’inibitore della piruvato deidrogenasi chinasi dicloroacetato (DCA).
RISULTATI
L’attivazione di RARβ riduce la crescita e il tasso metabolico dei melanociti
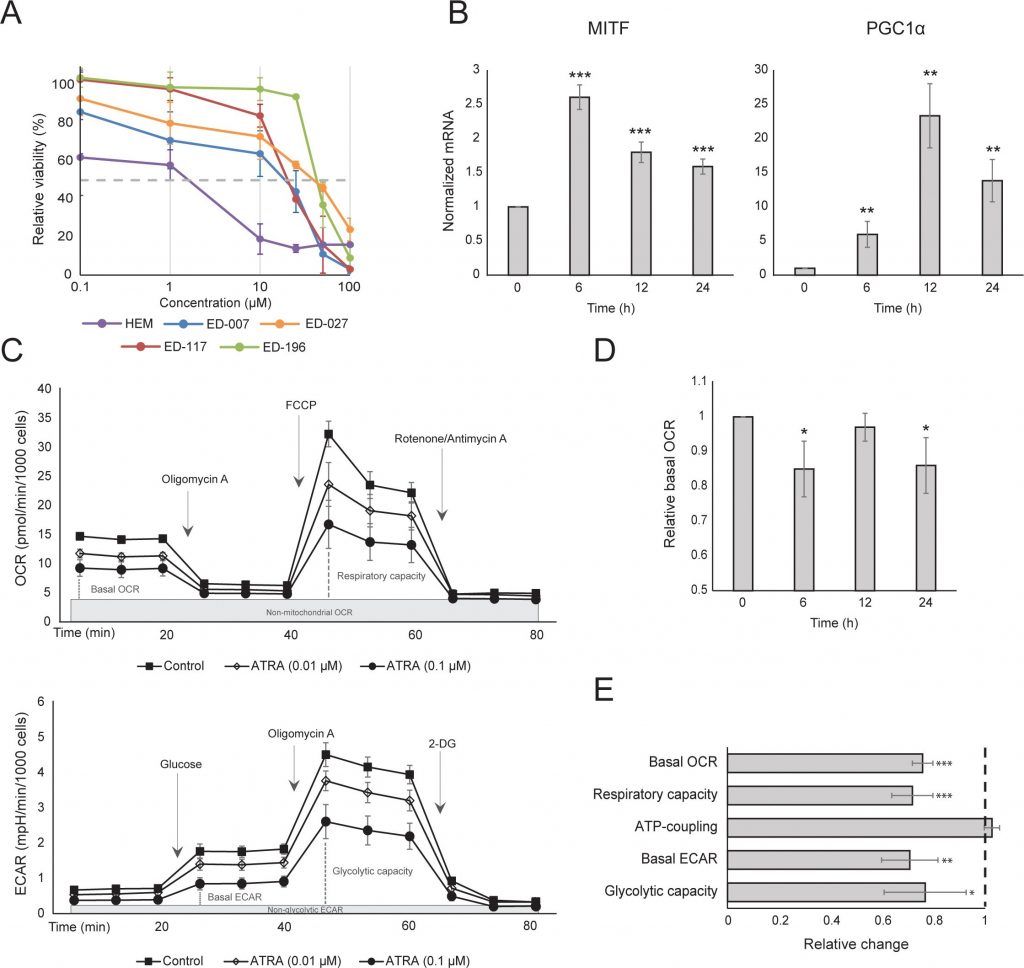
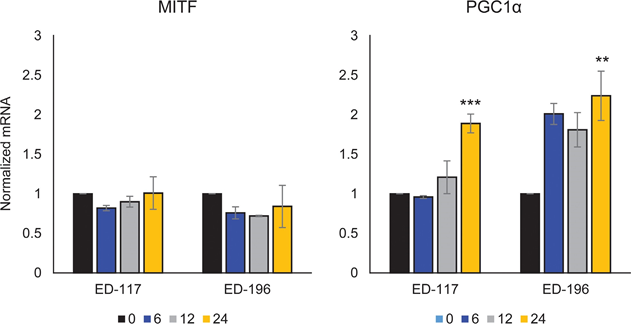
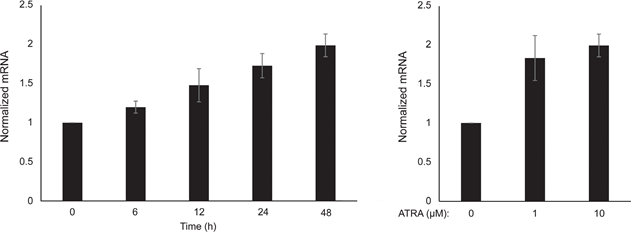
Abbiamo innanzitutto determinato l’effetto dell’attivazione di RARβ sulla crescita dei melanociti epidermici umani primari. Le cellule sono state trattate con l’agonista RARβ acido all-trans retinoico (ATRA) per 6 giorni e la risposta alla crescita è stata determinata con un saggio di vitalità basato sul cristalvioletto. Coerentemente con quanto riportato in precedenza [17, 20, 21], ATRA ha ridotto la crescita dei melanociti in modo dose-dipendente (Figura 1A), con un IC50 di 2,4 μM (Tabella 1). È stato precedentemente dimostrato che il trattamento a breve termine (<24 ore) con ATRA induce la differenziazione e la melanogenesi nei melanociti, mentre l’esposizione a lungo termine (>24 ore) riduce la proliferazione e induce l’apoptosi [20, 21]. Abbiamo riscontrato che l’ATRA (0,1 μM) ha indotto un’up-regolazione transitoria del fattore di trascrizione MITF (microphthalmia-associated transcription factor), specifico del lignaggio melanocitario, con un picco di espressione dopo 6 ore e un successivo declino verso i livelli basali (Figura 1B). Nelle cellule di melanoma, MITF regola l’espressione di PGC1α, un marcatore del fenotipo ossidativo [22]. Abbiamo quindi studiato l’espressione di PGC1α nei melanociti in diversi momenti dopo l’esposizione ad ATRA (0,1 μM). Come mostrato nella Figura 1B, anche PGC1α è stato transitoriamente upregolato, con un ritardo di circa 6 ore rispetto a MITF.
| Cellule/linee cellulari | Cellule/linee cellulari | Caratteristiche | Caratteristiche | Caratteristiche | Valori IC50 | Valori IC50 | Valori IC50 | Valori IC50 |
|---|---|---|---|---|---|---|---|---|
| Numero di ED | Nome | Stato di BRAF* | Espressione di RARβ ** | espressione di p14ARF | ATRA (μM) | LE135 (μM) | DCA (mM)*** | PLX4032 (μM) |
| HEM# | WT | + | + | 2.4±1.6 | 2.8±0.8 | 69.1±6.4 | NA | |
| ED-007 | FM-3 | WT | + | – | 18.6±8.7 | 8.6±1.0 | 12.2±2.2 | NA |
| ED-027 | FM-82 | BRAFV600E | + | + | 39.8±5.3 | 10.7±1.3 | 17.7±2.1 | 0.52±0.04 |
| ED-117 | Mel-NT3-00 | BRAFV600E | + | + | 25.5±5.0 | NA | 37.6 ±2.2 | 0.51±0.09 |
| ED-196 | Ma-Mel-51 | BRAFV600E | + | + | 46.2±9.1 | 8.4±0.4 | 35.8±3.2 | 0.26±0.06 |
I valori IC50 rappresentano la media ± la deviazione standard di ≥3 esperimenti indipendenti.
*Confermatodal pirosequenziamento
**Confermatodalla qPCR
***ValoriIC50 pubblicati da Abildgaard et al. [29]
#Melanociti epidermici umani
Dato il ruolo di PGC1α nella biogenesi mitocondriale, abbiamo poi indagato se l’espressione di PGC1α fosse correlata al livello di respirazione mitocondriale. Utilizzando lo strumento Seahorse XFe96, abbiamo misurato il tasso di consumo di ossigeno (OCR) e il tasso di acidificazione extracellulare (ECAR), indicatori rispettivamente del tasso respiratorio mitocondriale e dell’attività glicolitica. OCR ed ECAR sono stati misurati durante l’aggiunta sequenziale di modulatori metabolici, consentendo di determinare i tassi e le capacità basali dei due sistemi energetici (Figura 1C). Per comprendere la dipendenza temporale delle risposte all’ATRA, i parametri metabolici sono stati misurati dopo esposizioni a breve (6-24 ore) e a lungo termine (7 giorni). Dopo il trattamento dei melanociti con ATRA (0,1 μM) per 6 o 24 ore, l’OCR basale si è ridotto. Tuttavia, dopo 12 ore di trattamento, l’OCR era simile ai livelli basali (Figura 1D). Queste fluttuazioni nello stato metabolico hanno coinciso con i cambiamenti nell’espressione di MITF e PGC1α. L’esposizione a lungo termine (7 giorni) a una bassa dose di ATRA (0,01 μM) ha provocato un’ulteriore diminuzione sia dell’OCR basale che della capacità respiratoria (Figura 1E).
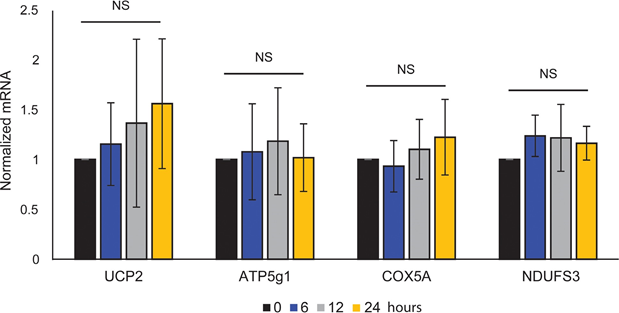
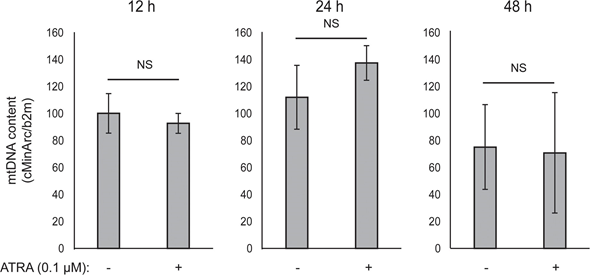
L’accoppiamento mitocondriale dell’ATP non è stato influenzato dall’ATRA (Figura 1E), con conseguente diminuzione netta della produzione di ATP da parte dei mitocondri. È stato dimostrato che la PGC1α regola la proteina di disaccoppiamento 2 (UCP2), determinando un leggero disaccoppiamento mitocondriale [23, 24]. L’espressione di UCP2 nei melanociti non è stata influenzata dal trattamento con ATRA (Figura 1 supplementare), a ulteriore sostegno del fatto che l’accoppiamento ATP è rimasto inalterato durante l’attivazione di RARβ. Anche l’espressione dei marcatori dell’attività mitocondriale (COX5A, ATP5g1 e NDUFS3) e il contenuto di DNA mitocondriale non sono stati influenzati dal trattamento con ATRA (0,1 μM) per un massimo di 24 e 48 ore, rispettivamente (Figure supplementari 1 e 2).
non ha mostrato alcuna significatività statistica (NS).
Il tasso glicolitico non ha subito variazioni dopo 24 ore di trattamento con ATRA (dati non mostrati); tuttavia, dopo 7 giorni, l’attività glicolitica basale e la capacità glicolitica erano significativamente ridotte (Figura 1E). La soppressione di entrambi i principali sistemi energetici cellulari indica che i melanociti presentano una minore richiesta di energia in presenza di ATRA, che potrebbe essere la conseguenza di una ridotta crescita cellulare.
L’inibizione di RARβ aumenta il tasso glicolitico basale e promuove la dipendenza glicolitica nei melanociti
Unasfida nello studio degli effetti cellulari dell’ATRA è la presenza di concentrazioni sconosciute di vitamina A nel siero fetale bovino, una fonte essenziale di micronutrienti nella maggior parte dei terreni di coltura cellulare [25]. Per studiare più in dettaglio il ruolo della segnalazione RARβ nel metabolismo dei melanociti, abbiamo quindi utilizzato l’antagonista RARβ LE135, che ha come bersaglio RARβ con moderata selettività rispetto a RARα ed elevata selettività rispetto a RARγ e RXRα [26].
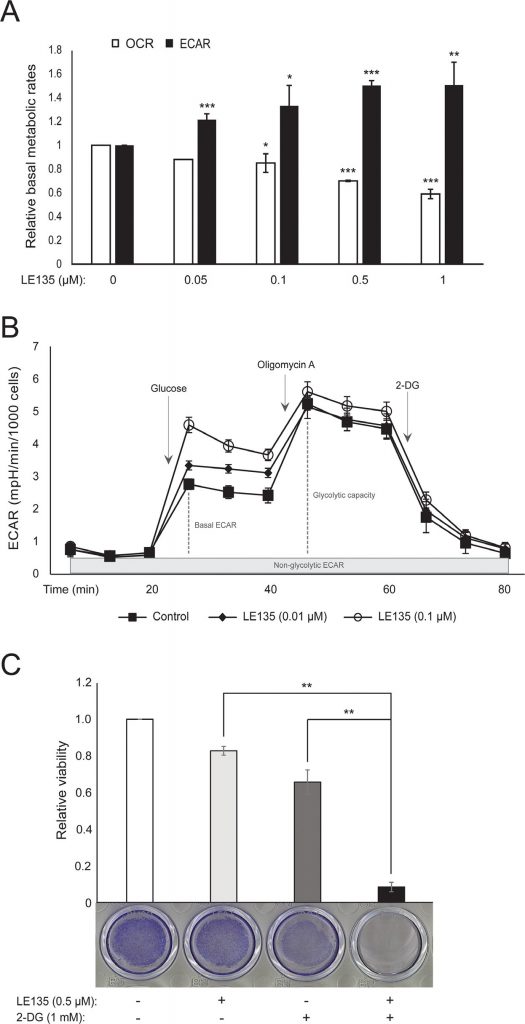
Abbiamo ripetuto i protocolli Seahorse mostrati nella Figura 1C su melanociti trattati con diverse concentrazioni di LE135 per 24 ore e 7 giorni. Dopo 24 ore, abbiamo osservato un aumento dose-dipendente dell’attività glicolitica, con un aumento dell’ECAR basale fino al 50% e una corrispondente riduzione dell’OCR (Figura 2A). L’ECAR basale era ancora aumentata dopo 7 giorni di trattamento (Figura 2B). Non si è registrato un aumento significativo della capacità glicolitica, il che suggerisce che le cellule sono state costrette a fare affidamento sulla glicolisi per la produzione di energia. Ciò è stato ulteriormente supportato da una maggiore sensibilità di queste cellule all’inibitore della glicolisi 2-deossi-D-glucosio (2-DG), in presenza di LE135 (Figura 2C).
L’ATRA antagonizza l’effetto dell’inibizione di BRAF nelle cellule di melanoma
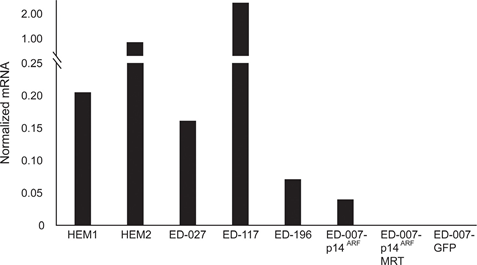
RARB è silenziato dall’ipermetilazione del promotore in molti melanomi, il che suggerisce che possiede proprietà soppressive del tumore [16]. In un precedente studio sugli eventi genetici ed epigenetici in 110 linee cellulari di melanoma, abbiamo riscontrato una prevalenza del 66% di mutazioni di BRAF e del 45% di ipermetilazione del promotore di RARB, senza alcuna correlazione tra questi due eventi [17]. Per ampliare questi dati, abbiamo esaminato l’espressione di RARβ in 84 di queste linee cellulari di melanoma e nei melanociti epidermici umani. I livelli di espressione variavano notevolmente tra le linee cellulari di melanoma, andando dalla completa assenza di espressione a livelli fino a 27 volte superiori a quelli dei melanociti (Figura 3A). Come previsto, l’ipermetilazione del promotore di RARB era associata a livelli di espressione di RARβ bassi o non rilevabili, con poche eccezioni. Non vi è stata alcuna associazione tra le mutazioni BRAFV600E e i livelli di espressione di RARβ (Figura 3A), suggerendo che la sensibilità delle cellule di melanoma all’ATRA può essere indipendente dallo stato di BRAF.
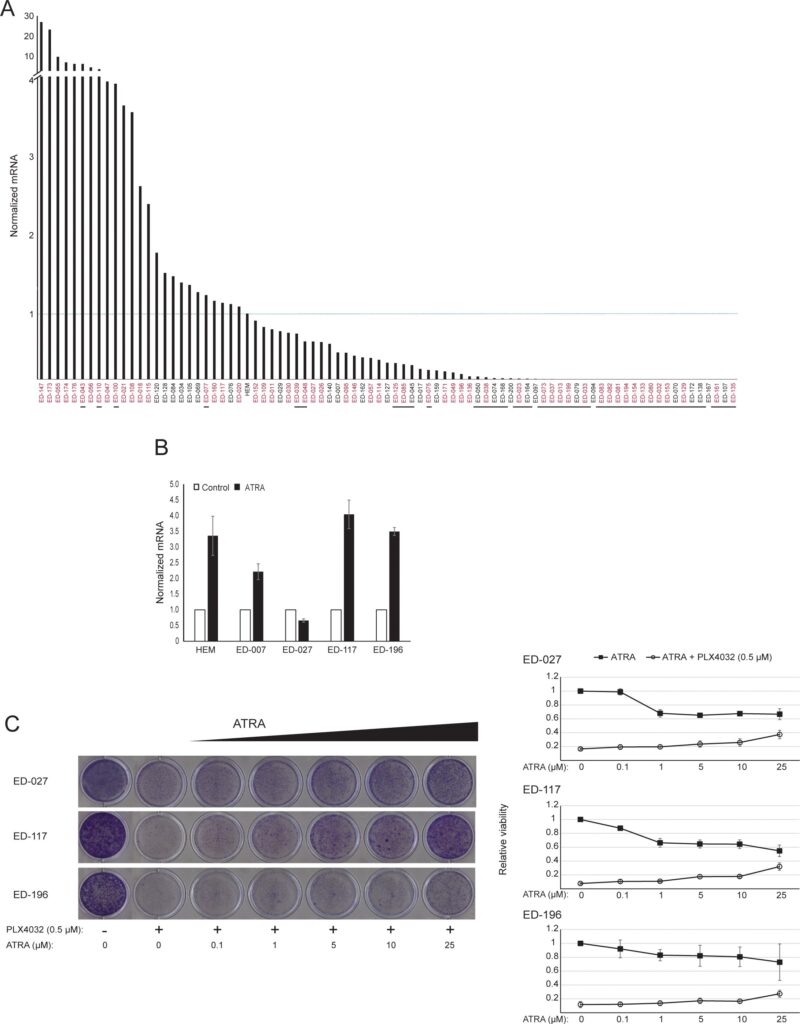
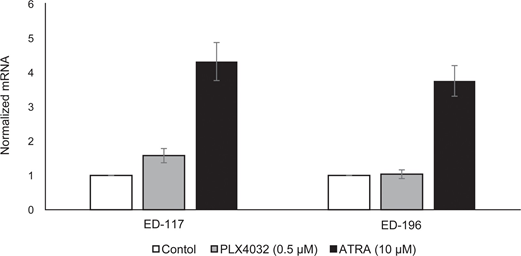
Per studiare ulteriormente il ruolo della segnalazione RARβ nel melanoma, abbiamo selezionato quattro linee cellulari di melanoma RARβ-positive (ED-007, ED-027, ED-117 e ED-196) per l’analisi funzionale. Tre di queste linee cellulari erano mutate BRAFV600E (ED-027, ED-117 e ED-196) e una era BRAF wild type (ED-007). Il trattamento con ATRA ha portato a una riduzione della crescita di tutte e quattro le linee cellulari (Figura 1A), sebbene la loro sensibilità fosse inferiore a quella dei melanociti, come indicato dai valori di IC50 (Tabella 1). È noto che l’espressione di RARβ è indotta in risposta ad ATRA [27]. Come mostrato nella Figura 3B, RARβ è stato indotto in 3 delle quattro linee cellulari di melanoma. Nonostante un’induzione di RARβ più pronunciata in ED-117 e ED-196, l’effetto di ATRA sull’espressione di MITF e PGC1α è stato attenuato rispetto ai melanociti (Figura 3 supplementare). La soppressione dell’asse MITF/PGC1α è stata precedentemente dimostrata come una conseguenza dell’attività oncogenica di BRAF [11], che potrebbe contribuire a una minore risposta all’ATRA. Coerentemente con questa idea, le cellule BRAF wild type hanno mostrato la maggiore sensibilità all’ATRA, anche se ancora notevolmente inferiore a quella dei melanociti (Tabella 1).
Per verificare se il targeting di BRAF con PLX4032 ripristinasse la sensibilità all’ATRA, abbiamo trattato le linee cellulari di melanoma BRAFV600E-mutanti con PLX4032 a una concentrazione vicina ai valori IC50 (cfr. Tabella 1) in combinazione con concentrazioni crescenti di ATRA (0,1-25 μM). È interessante notare che ATRA ha salvato l’effetto citotossico di PLX4032 in tutte le linee cellulari. L’effetto dose-dipendente di ATRA sulla crescita delle cellule di melanoma trattate con PLX4032 (Figura 3C) indica un antagonismo tra i due composti. Il trattamento con PLX4032 (0,5 μM) non ha ridotto l’espressione di RARβ (Figura 4 supplementare), suggerendo un meccanismo diverso per questo antagonismo.
L’effetto dell’ATRA sul metabolismo cellulare è influenzato dallo stato di p14ARF
L’osservazione che sia l’ATRA che PLX4032 influenzano la biogenesi mitocondriale potrebbe indicare una spiegazione metabolica per l’effetto antagonista di questi composti. In precedenza è stato dimostrato che p14ARF è espresso come proteina citoplasmatica nei melanociti normali e che protegge queste cellule da mitocondri disfunzionali [19]. In accordo con i risultati precedenti [17], l’ATRA ha aumentato l’espressione di p14ARF nelle cellule di melanoma RARβ-positive (Figura 5 supplementare). Paradossalmente, sebbene p14ARF sia spesso perso nel melanoma attraverso la delezione del locus CDKN2A, non è possibile abbattere stabilmente ARF nelle cellule di melanoma che esprimono questo gene [17]; e dati non mostrati). Invece, per studiare il ruolo potenziale di p14ARF nel mediare la risposta cellulare all’ATRA, abbiamo trasfettato in modo stabile la linea cellulare di melanoma ED-007 carente di p14ARF con un costrutto EGFP-p14ARF. L’espressione di p14ARF nelle cellule trasfettate è stata verificata con qPCR (Figura 6 supplementare). L’analisi Seahorse ha mostrato profili metabolici diversi (Figura 4A) con un OCR e una capacità respiratoria basale significativamente più bassi nelle cellule con espressione p14ARF ripristinata rispetto alle cellule trasfettate di controllo (Figura 4B). È interessante notare che le cellule che esprimono p14ARF mostrano anche una maggiore sensibilità all’ATRA (Figura 4C).
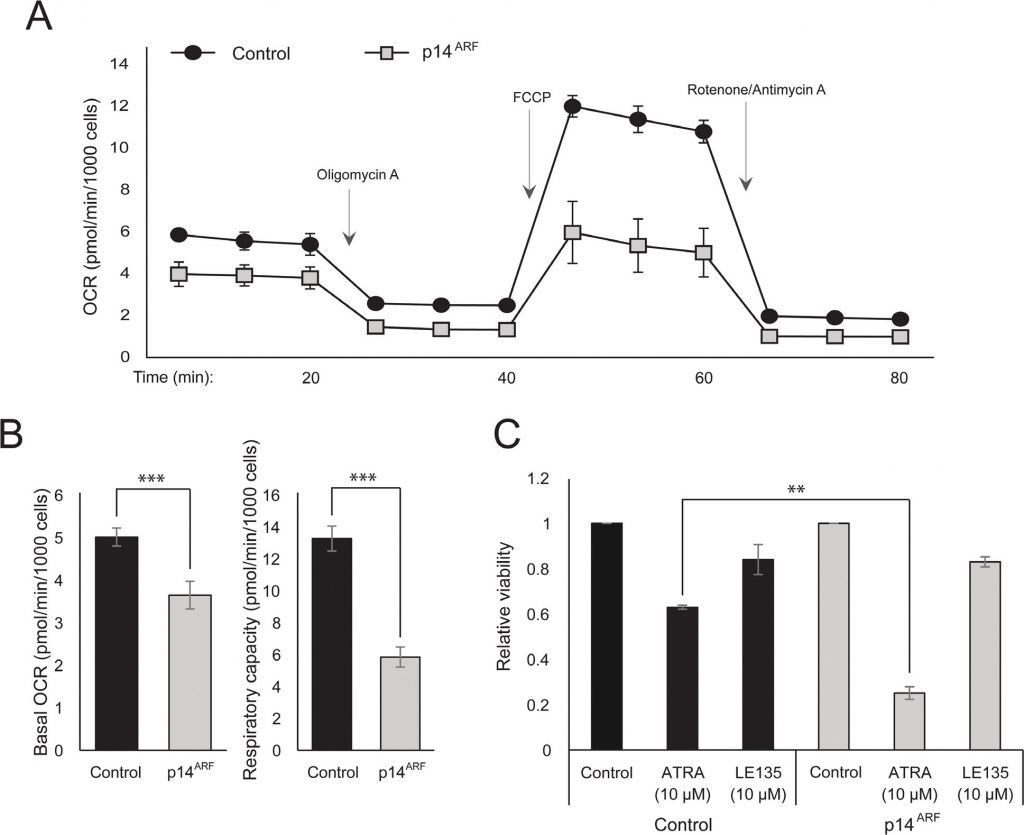
Ilblocco di RARβ induce dipendenza glicolitica e stress energetico nelle cellule di melanoma e le sensibilizza al dicloroacetato
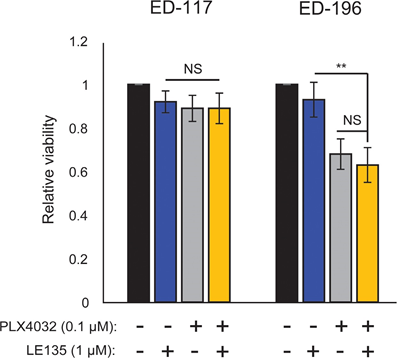
Sulla base della scoperta di un effetto antagonista tra PLX4032 e ATRA, abbiamo testato l’effetto combinato di PLX4032 e LE135 per verificare il potenziale sinergismo nelle cellule di melanoma ED-117 e ED-196. Non è stata dimostrata alcuna inibizione cooperativa della crescita del melanoma nella configurazione sperimentale qui utilizzata (6 giorni di trattamento con PLX4032 [0,1 μM] e LE135 [1 μM]; Figura supplementare 7).
Per studiare ulteriormente l’effetto dell’inibizione di RARβ sul metabolismo del melanoma, abbiamo misurato OCR ed ECAR in linee cellulari di melanoma RARβ-positive trattate con LE135. Analogamente a quanto osservato nei melanociti (Figura 2A-2B), LE135 ha aumentato il tasso glicolitico basale e ridotto la respirazione mitocondriale basale nelle cellule di melanoma (Figura 5A-5B). Questo spostamento metabolico, coerente con l’effetto Warburg, è stato più netto nelle cellule di melanoma rispetto ai melanociti, portando a un aumento del tasso glicolitico basale per raggiungere la capacità massima. Ciò è stato dimostrato dall’incapacità di aumentare ulteriormente l’ECAR dopo l’aggiunta di oligomicina A, indicando una mancanza di flessibilità metabolica (Figura 5B). Per studiare l’effetto di LE135 sulla bioenergetica cellulare, abbiamo analizzato lo stato di fosforilazione dell’AMP-activated protein kinase (AMPK). L’AMPK rileva lo stato energetico cellulare reagendo ad alti livelli di AMP, che si accumula quando il rapporto ATP/ADP diminuisce. Pertanto, la riduzione della produzione di energia o l’aumento del consumo energetico possono promuovere un aumento dell’AMP, che si lega all’AMPK e porta alla sua fosforilazione e attivazione [28]. Il trattamento delle linee cellulari di melanoma con LE135 ha portato alla fosforilazione di AMPK, indicando che le cellule sono sottoposte a stress energetico. L’aumento dei livelli di p-AMPK è stato evidente già dopo 2 ore ed è ulteriormente aumentato fino ad almeno 48 ore (Figura 5C). Questi risultati sono diversi da quelli ottenuti nei melanociti, dove il trattamento con LE135 non ha portato all’attivazione di AMPK (Figura 8 supplementare). Sia le cellule di melanoma che i melanociti hanno mostrato una riduzione della crescita cellulare durante 6 giorni di trattamento con LE135 (i valori IC50 sono riportati nella Tabella 1). Inoltre, analogamente ai melanociti, LE135 ha sensibilizzato le cellule di melanoma all’inibizione glicolitica con 2-DG (Figura 5D).
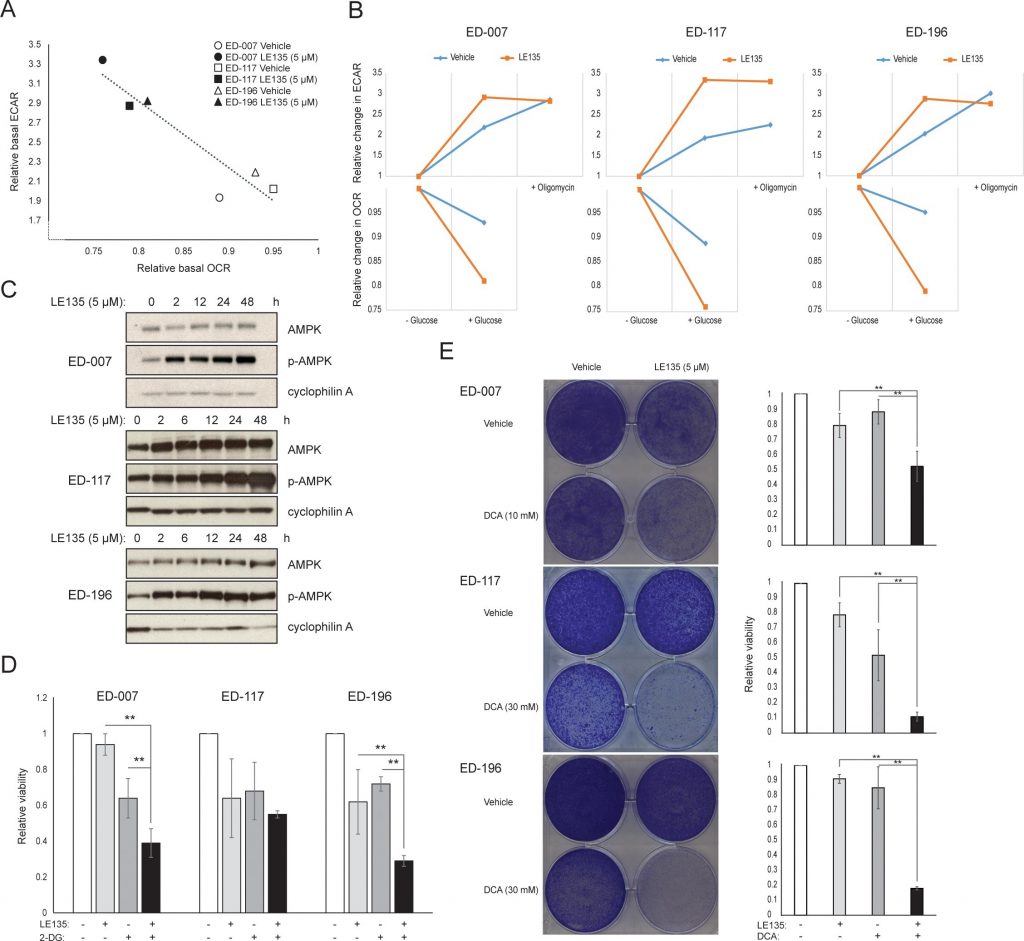
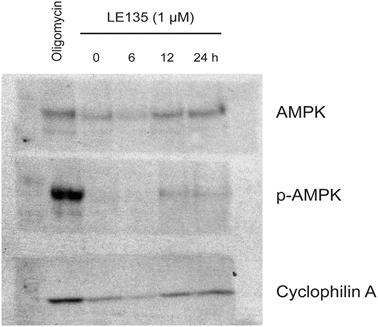
L’induzione dello stress energetico con LE135 nelle cellule di melanoma ma non nei melanociti allude a una potenziale rilevanza terapeutica di questo composto nelle strategie di trattamento combinato. L’inibitore della piruvato deidrogenasi chinasi DCA ha precedentemente dimostrato di inibire la crescita delle cellule di melanoma inducendo uno spostamento del metabolismo dalla glicolisi, rendendo le cellule dipendenti dalla respirazione mitocondriale [9,29-32]. Inoltre, è stato dimostrato che il DCA inibisce la crescita di una serie di linee cellulari di melanoma, indipendentemente dallo stato di BRAF e dalla sensibilità al PLX4032 [29]. Per studiare l’effetto combinato di LE135 e DCA, abbiamo applicato concentrazioni inferiori ai rispettivi valori IC50 per ciascuna delle tre linee cellulari di melanoma (cfr. Tabella 1). Nonostante un basso effetto sulla riduzione della crescita di ciascun composto singolarmente (9-21% per LE135 e 12-48% per DCA), la combinazione ha indotto una riduzione fino all’89% (Figura 5E). Questi risultati suggeriscono che gli effetti opposti di LE135 (che promuove la dipendenza glicolitica) e DCA (che allontana le cellule dalla glicolisi) possono agire sinergicamente per inibire la crescita del melanoma.
DISCUSSIONE
L’ATRA e altri derivati della vitamina A riducono la crescita cellulare e inducono l’espressione di marcatori di differenziazione in vari tessuti [33, 34]. Abbiamo riscontrato che, nei melanociti umani primari, l’ATRA induce una transitoria upregulation dell’asse MITF/PGC1α, coerentemente con l’aumento della funzione mitocondriale indotto dall’ATRA osservato in altri tipi di cellule come gli adipociti e gli epatociti [35-37]. Tuttavia, il trattamento a lungo termine con basse concentrazioni di ATRA ha portato a una riduzione della crescita cellulare e del tasso metabolico, determinata da una minore glicolisi basale e da una minore respirazione mitocondriale. Questi cambiamenti metabolici in risposta all’ATRA riflettono probabilmente una risposta di differenziazione verso lo stato non proliferativo che caratterizza i melanociti residenti nella pelle. Il blocco della segnalazione di RARβ in queste cellule ha determinato un aumento del tasso glicolitico basale e una corrispondente diminuzione del metabolismo ossidativo. Il vantaggio selettivo della perdita della funzione di RARβ nel melanoma, ad esempio attraverso l’ipermetilazione di RARB, potrebbe essere correlato alla transizione verso un fenotipo più dipendente dalla glicolisi che supporta l’effetto Warburg.
Contrariamente a quanto accade nei melanociti primari, il blocco della segnalazione di RARβ nelle cellule di melanoma ha portato a uno stress energetico, come indicato dall’attivazione di AMPK. Questa risposta potrebbe essere il risultato di una minore capacità delle cellule di melanoma di aumentare la glicolisi rispetto ai melanociti. Mentre i melanociti hanno un livello glicolitico basale relativamente basso e possono passare a un’attività più elevata quando necessario, le cellule di melanoma sono caratterizzate da un tasso glicolitico elevato vicino alla loro capacità massima. Pertanto, i melanociti sono più flessibili nell’adattarsi all’inibizione del segnale RARβ per sostenere il livello energetico. Ciò indica che esiste una finestra terapeutica rilevante per LE135 e altri inibitori di RARβ in terapie combinate, ad esempio con DCA. Analogamente agli effetti metabolici dell’inibizione di BRAF, il DCA fa passare le cellule tumorali glicolitiche dalla glicolisi alla respirazione mitocondriale [9, 38, 39], ma a differenza del PLX4032, l’effetto del DCA non è limitato ai melanomi BRAF-mutati [29]. In uno studio precedente, abbiamo dimostrato che il cambiamento metabolico indotto dal DCA era correlato a una riduzione dei livelli di ATP, suggerendo che il DCA potrebbe colpire l’omeostasi bioenergetica delle cellule di melanoma [29]. Qui abbiamo scoperto che la combinazione di LE135 e DCA ha attenuato in modo cooperativo la crescita delle cellule di melanoma che esprimono il recettore RARβ. Il trattamento delle cellule di melanoma con DCA o LE135 potrebbe dare loro una finestra per adattarsi alle nuove esigenze metaboliche, mentre la combinazione dei trattamenti limiterebbe la flessibilità metabolica e le renderebbe incapaci di sostenere la produzione di energia necessaria per una crescita continua.
In molti melanomi, l’espressione di PGC1α è bassa a causa della soppressione di MITF da parte delle mutazioni oncogeniche di BRAF [11]. Questo fenotipo supporta l’effetto Warburg, costringendo le cellule a passare alla glicolisi per la produzione di energia. Il trattamento con inibitori di BRAF ripristina l’espressione di PGC1α e riporta il modello metabolico verso la respirazione mitocondriale [11, 40]. L’effetto citotossico dell’inibizione di BRAF può essere potenziato dalla presenza di mitocondri disfunzionali nelle cellule di melanoma, con conseguente aumento della produzione di ROS [13, 41]. Abbiamo scoperto che ATRA aumenta la crescita delle cellule di melanoma in presenza dell’inibitore di BRAF PLX4032. Sulla base delle conoscenze di studi precedenti [11, 17, 19, 41] e dei risultati qui presentati, proponiamo un modello che integra gli effetti metabolici di PLX4032 e ATRA e che spiega il loro antagonismo (Figura 6). Il modello suggerisce un duplice ruolo dell’aumento della segnalazione RARβ, che porta all’attivazione di PGC1α e alla biogenesi mitocondriale, mentre allo stesso tempo sopprime l’OCR e la produzione di ROS attraverso l’induzione di p14ARF. Il modello è stato supportato dai risultati del presente studio e di uno precedente [17] che dimostrano che il trattamento con ATRA induce l’espressione di p14ARF e che la ricostituzione delle cellule di melanoma p14ARF-deficienti con p14ARF wild-type abbassa l’OCR e aumenta la sensibilità all’ATRA. Pertanto, l’ATRA potrebbe ridurre la sensibilità dei melanomi RARβ-positivi al PLX4032 limitando la citotossicità derivante dalla produzione di ROS. La vitamina A è stata proposta per scopi profilattici e terapeutici in molti tipi di cancro, compreso il melanoma [42]. Sebbene manchi una validazione clinica, i nostri risultati sconsigliano l’uso della supplementazione di vitamina A nei pazienti con melanoma in trattamento con inibitori di BRAF, a causa del potenziale effetto antagonizzante.
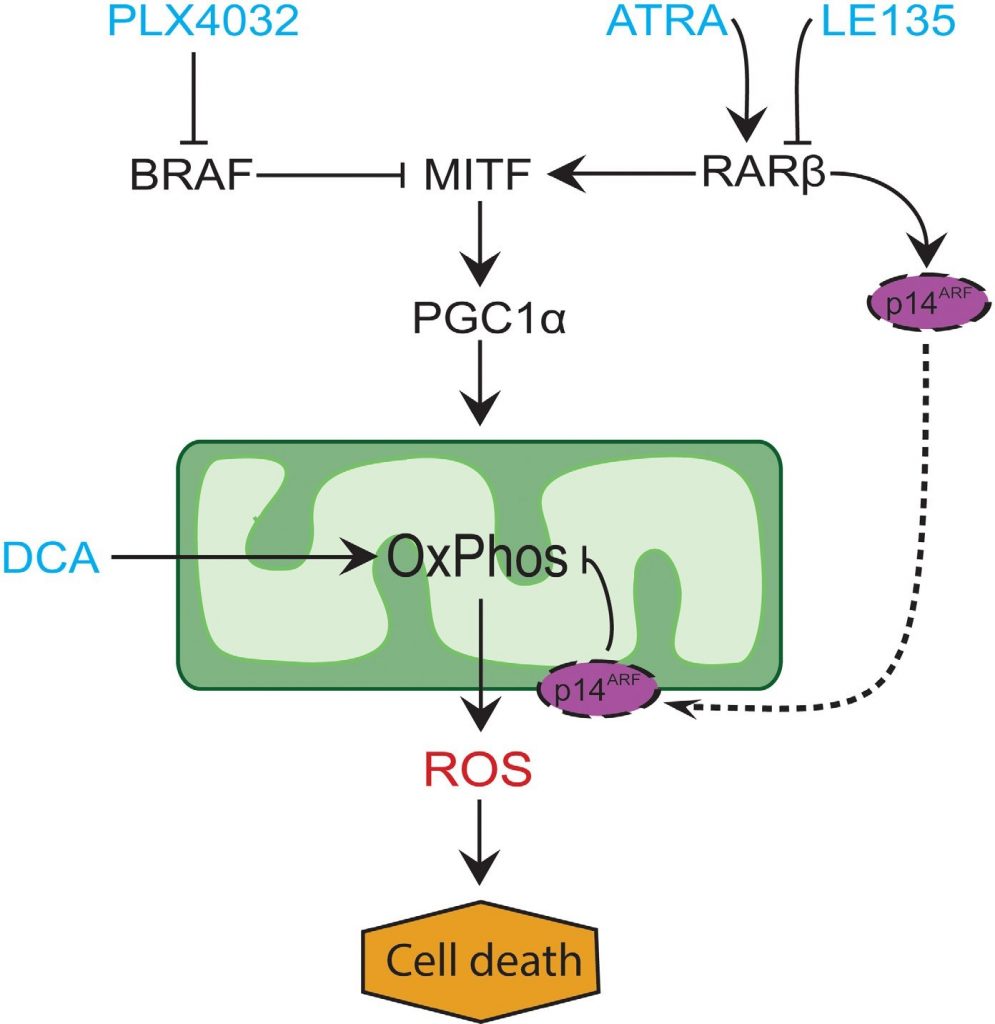
In conclusione, abbiamo identificato una nuova funzione della segnalazione di RARβ nel metabolismo delle cellule melanocitarie e del melanoma, che potrebbe avere implicazioni cliniche. La capacità di RARβ di attivare la via MITF-PGC1α, e potenzialmente una riduzione dell’attività respiratoria mitocondriale dipendente da p14ARF, influisce negativamente sulla risposta terapeutica all’inibizione di BRAF. Tuttavia, il blocco della segnalazione di RARβ promuove la dipendenza glicolitica nelle cellule di melanoma e potenzia l’effetto del DCA, che potrebbe essere sfruttato a livello terapeutico.
MATERIALI E METODI
Reagenti
Sodio dicloroacetato (DCA), 2-deossi-D-glucosio (2-DG), acido retinoico all-trans (ATRA) e LE135 sono stati acquistati da Sigma-Aldrich. Il DCA e il 2-DG sono stati disciolti in dH2Oa una concentrazione di base di 1 M. L’ATRA e il LE135 sono stati disciolti in DMSO a una concentrazione di base di 0,1 M. PLX4032 (vemurafenib) è stato acquistato da Selleck Chemicals e disciolto in DMSO a una concentrazione di base di 0,05 M.
Coltura cellulare
Le linee cellulari di melanoma sono state ottenute dall’European Searchable Tumour line Database (ESTDAB, ED) [43]. Lo stato di queste linee cellulari rispetto alle mutazioni di BRAF e alla metilazione del promotore di RARB è stato descritto in precedenza [17]. I melanociti epidermici umani primari (neonatali) provenienti da tessuto leggermente pigmentato (HEMn-LP; denominati melanociti) sono stati acquistati da Invitrogen (C0025C). Per gli esperimenti sono stati utilizzati melanociti provenienti da tre diversi individui (lotto n. 200706893 da marzo 2015 e novembre 2016; lotto n. 1583282 da febbraio 2017). Le linee cellulari di melanoma sono state coltivate a 37°C al 5% diCO2 in terreno RPMI-1640 integrato con il 10% di siero fetale bovino. Le cellule HEMn-LP sono state coltivate nelle stesse condizioni in terreno 254CF integrato con l’1% di supplemento per la crescita dei melanociti umani (HMGS), incluso il forbolo 12-miristato 13-acetato (PMA). Per i setup sperimentali, le cellule HEMn-LP sono state coltivate in terreno integrato con HMGS-2 (senza PMA). Tutti i terreni e i supplementi sono stati acquistati da Invitrogen.
Analisi della vitalità cellulare
Per valutare l’effetto dei composti studiati sulla vitalità cellulare è stato applicatoilsaggio del violetto di cristallo. Le cellule sono state seminate in duplicato e trattate con i composti in questione o con il veicolo di controllo per 6 giorni. Il terreno e i composti di trattamento sono stati sostituiti ogni 48 ore. Gli esperimenti sono stati ripetuti tre volte in modo indipendente. Dopo il periodo di trattamento, il terreno e le cellule non attaccate sono stati rimossi, mentre le cellule rimanenti sono state lavate in PBS e fissate con glutaraldeide per 15 minuti. Le cellule fissate sono state incubate con una soluzione di cristalvioletto (0,1% cristalvioletto, 20% CH3OH) per 1 ora. La quantità di colorante assorbita dal monostrato, proporzionale al numero di cellule vitali attaccate al fondo del pozzetto, è stata quantificata estraendo il colore con acido acetico al 10% e misurando l’assorbanza a una lunghezza d’onda di 595 nm. La vitalità relativa dopo il trattamento con ATRA, LE135 o PLX4032 è stata utilizzata per determinare la concentrazione inibitoria semimassimale (IC50). Tracciando la curva dose-risposta, il valore IC50 è stato stimato come la concentrazione al punto di vitalità cellulare del 50%.
Purificazione del DNA e dell’RNA
Il DNA per la quantificazione del mtDNA e l’RNA per la sintesi del cDNA sono stati purificati simultaneamente con AllPrep DNA/RNA/Protein mini kit (Qiagen) secondo il protocollo fornito.
Analisi dell’espressione
La sintesi di cDNA è stata eseguita con qScript™ XLT cDNA SuperMix (Quanta Bioscience). L’espressione genica di PGC1α, MITF, RARβ, p14ARF, UCP2, ATP5g1, COX5A e NDUFS3 è stata determinata con la PCR quantitativa in tempo reale su Roche LightCycler 2.0 utilizzando il kit LightCycler FastStart DNA MasterPLUS SYBR Green I (Roche). I primer sono elencati nella Tabella supplementare 1.
| Gene | Primer in avanti | Primer inverso |
| PGC1α* | GTAAATCTGCGGGATGATGG | AATTGCTTGCGTCCACAAA |
| MITF** | CCGTCTCACTGGATTGGT | TACTTGGTGGGGTTCGAG |
| p14ARF*** | CCCTCGTGCTGATGCTACTGA | CATGACCTGGTCTTCTAGGAAGC |
| RARβ*** | TCCTGGATTTCTACACTGCG | AAGCAGGGTTTGTACACTCG |
| UCP2 | AAGACCATTGCCCGAGAGG | TTGGTTCAGGAGGGCAT |
| ATP5g1* | ATCATTGGCTATGCCAGGAA | ATGGCGAAGAGGATGAGGA |
| COX5A* | GGGAATTGCGTAAAGGGATAA | TCCTGCTTTGTCCTTAACAACC |
| NDUFS3* | GCTGACGCCCATTGAGTCTG | GGAATTTGGGCCAACTCC |
| RPLP0 | ACTAAAATCTCCAGGGGCACC | ATGACCAGCCCAAAGGAGAA |
*Seguenze di primer pubblicate da Vazquez et al. 2013 [22].
**Seguenze di primer pubblicate da Haq et al. 2014 [11].
***Seguenze di primer pubblicate da Dahl et al. 2013 [17].
Immunoblotting
I campionisono stati preparati da fiasche di coltura cellulare con tampone di lisi (SLB) integrato con β-mercaptoetanolo incolore (BPB), Phospho-Stop e inibitore della proteasi (Thermo Fisher Scientific). I lisati cellulari sono stati ripuliti mediante centrifugazione a 20.000 rpm per 3 minuti. La concentrazione proteica è stata misurata con il Qubit Protein Assay Kit (Thermo Fisher Scientific) e 50 μg di proteine di ciascun campione sono stati caricati su un gel SDS a 10 pozzetti, Bis-Tris NuPage al 4-12% (Invitrogen). Le proteine sono state separate a 80 V per 30 minuti, seguiti da 110 V fino al completamento. Il blotting è stato eseguito con un’unità di trasferimento semisecca su una membrana di nitrocellulosa ECL a 3,3 mA/1 cm2/1 h/gel. Successivamente la membrana è stata colorata con Ponceau. La membrana è stata bloccata in latte al 5% per 1 ora, quindi lavata due volte per 5 minuti con TBST e colorata con anticorpi anti-AMPK o anti-p-AMPK (Thr172) (Cell Signaling; 1:2000) in BSA al 5% a 4°C e con un anticorpo anti-ciclofilina A (Cell Signaling; 1:5000) come controllo di carico. Dopo tre cicli di lavaggio di 10 minuti con TBST, la membrana è stata colorata con l’anticorpo secondario (anti-rabbit; DakoCytomation; 1:2000) per 1 ora a temperatura ambiente, seguito da altri 3 cicli di lavaggio. Le proteine sono state visualizzate utilizzando il substrato ECL Plus Western Blotting (Thermo Fisher Scientific) 1:1 per 2-3 minuti.
Analisi metabolica
L’analisi metabolica è stata eseguita su linee cellulari di melanoma e melanociti utilizzando un analizzatore Seahorse XFe96 (Seahorse Bioscience, Billerica, MA), che esegue misurazioni in tempo reale del tasso di acidificazione extracellulare (ECAR) e del tasso di consumo di ossigeno (OCR). Le cellule sono state seminate a 20.000 per pozzetto in micropiastre per colture cellulari Seahorse 24 ore prima di eseguire le misurazioni. Le variazioni dell’attività basale e della capacità dei sistemi energetici mitocondriale e glicolitico sono state determinate utilizzando il Mito Stress Test Kit e il Glycolysis Stress Test Kit (Agilent Technologies). I test sono stati eseguiti secondo i protocolli forniti. Il Mito Stress Test è stato eseguito nel normale terreno di coltura, mentre nel test di stress della glicolisi il terreno è stato sostituito con terreno Seahorse XF Base integrato con L-glutammina (2 mM), con pH regolato a 7,4, 1 ora prima delle misurazioni. Per esposizioni più lunghe (>24 ore), le cellule sono state trattate in fiasche di coltura prima della semina. Tutti i risultati sono stati normalizzati al numero di cellule seminate, poiché le concentrazioni di ATRA e LE135 utilizzate non hanno influenzato la crescita cellulare durante le 24 ore. Il protocollo per l’esecuzione dei saggi nella macchina Seahorse prevedeva cicli di 3 minuti di miscelazione/3 minuti di misurazione. Sono stati eseguiti tre esperimenti indipendenti con 6 repliche di ciascun campione.
Trasfezione
La linea cellulare di melanoma ED-007 è stata trasfettata con vettori di espressione pEGFP (controllo) o pEGFP-p14ARF (2 μg di vettore/2 ×106 cellule), entrambi contenenti GFP come gene reporter. I costrutti sono stati ottenuti come precedentemente descritto [19]. La trasfezione è stata eseguita utilizzando la tecnologia di nucleofezione Amaxa, buffer V, programma T-020, seguendo il protocollo raccomandato dal produttore. Il successo della trasfezione è stato verificato visivamente. I cloni stabili sono stati selezionati utilizzando 400 μg/ml di G418 (geneticina; Thermo Fisher Scientific). Nella configurazione sperimentale, le cellule sono state seminate senza G418.
Quantificazione del DNA mitocondriale
Il DNAmitocondrialeè stato quantificato mediante reazione a catena della polimerasi digitale a goccia (ddPCR) utilizzando il sistema QX200 (BioRad Laboratories, Hercules, CA, USA). Per ogni reazione sono stati utilizzati circa 0,5 ng di DNA. Il numero di copie mitocondriali è stato determinato calcolando il rapporto tra un sito di DNA mitocondriale (mtMinArc) e un locus nucleare a singola copia (β2m) come descritto da Phillips et al. [44]. I primer, le sonde e le condizioni sperimentali sono elencati nella Tabella 2 supplementare.
| mtMinArc | β2m | |
| Primer in avanti* | CTAAATAGCCCACGTTCCC | GCTGGGTAGCTCTAAACAATGTATTCA |
| Primer inverso* | AGAGCTCCCGTGAGTGGTTA | CCATGTACTAACAAATGTCTAAAATGGT |
| Sonda* | 6FAM-CATCACGATGGATCACAGGT(NFQ) | VIC-CAGCAGCCTATTCTGC(NFQ) |
| Conc. primer | 75 nM | 500 nM |
| Temperatura di ricottura | 50°C | 52°C |
| Numero di cicli | 40 | 40 |
*Seguenze del primer e della sonda pubblicate da Phillips et al. [44].
Analisi statistica
Le differenze tra serie di dati indipendenti sono state determinate con il test t di Student. Per l’analisi statistica della varianza tra i diversi trattamenti è stata utilizzata l’ANOVA a campioni appaiati a una via. Per determinare la significatività statistica è stato utilizzato il test di confronto multiplo HSD (honest significance difference) di Tukey.
Contributi degli autori
CA e PG hanno pianificato e organizzato lo studio. CA ha eseguito la maggior parte degli esperimenti e l’elaborazione dei dati, compresi la coltura cellulare, i protocolli di trattamento, l’analisi metabolica e le statistiche. CD ha pianificato ed eseguito la trasfezione di EGFP-p14ARF e le misurazioni del mtDNA e ha contribuito all’interpretazione dei risultati. AA ha eseguito e ottimizzato i protocolli di immunoblotting e PCR quantitativa. AC ha eseguito i saggi di coltura cellulare. CA e PG hanno scritto il manoscritto con contributi e modifiche di CD, AA e AC. Il manoscritto finale è stato letto e approvato da tutti gli autori.
CONFLITTI DI INTERESSE
Gli autori dichiarano di non avere conflitti di interesse.
FINANZIAMENTO
Questo studio è stato sostenuto dalla Danish Cancer Society
RIFERIMENTI
1 Tassi di sopravvivenza per il tumore cutaneo del melanoma, per stadio. (cancer.org: American Cancer Society).2 Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, Chodon T, Nelson SF, McArthur G, et al. I melanomi acquisiscono resistenza all’inibizione di B-RAF (V600E) attraverso l’upregulation di RTK o N-RAS. Nature. 2010; 468: 973-7. https://doi.org/10.1038/nature09626.
3 Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP, Barretina J, Caponigro G, Hieronymus H, Murray RR, et al. COT guida la resistenza all’inibizione di RAF attraverso la riattivazione della via delle MAP chinasi. Nature. 2010; 468: 968-72. https://doi.org/10.1038/nature09627.
4 Miller AJ, Mihm MC Jr. Melanoma. N Engl J Med. 2006; 355: 51-65. https://doi.org/10.1056/NEJMra052166.
5 Abildgaard C, Guldberg P. Molecular drivers of cellular metabolic reprogramming in melanoma. Trends Mol Med. 2015; 21: 164-71. https://doi.org/10.1016/j.molmed.2014.12.007.
6 Ratnikov BI, Scott DA, Osterman AL, Smith JW, Ronai ZA. Ricablaggio metabolico nel melanoma. Oncogene. 2017; 36: 147-57. https://doi.org/10.1038/onc.2016.198.
7 Warburg O, Wind F, Negelein E. Il metabolismo dei tumori nell’organismo. J Gen Physiol. 1927; 8: 519-30.
8 Hall A, Meyle KD, Lange MK, Klima M, Sanderhoff M, Dahl C, Abildgaard C, Thorup K, Moghimi SM, Jensen PB, Bartek J, Guldberg P, Christensen C. La fosforilazione ossidativa disfunzionale rende le cellule di melanoma maligno dipendenti dalla glicolisi guidata dall’oncogene BRAF (V600E). Oncotarget. 2013; 4: 584-99. https://doi.org/10.18632/oncotarget.965.
9 Parmenter TJ, Kleinschmidt M, Kinross KM, Bond ST, Li J, Kaadige MR, Rao A, Sheppard KE, Hugo W, Pupo GM, Pearson RB, McGee SL, Long GV, et al. La risposta del melanoma BRAF-mutante all’inibizione di BRAF è mediata da una rete di regolatori trascrizionali della glicolisi. Cancer Discov. 2014; 4: 423-33. https://doi.org/10.1158/2159-8290.CD-13-0440.
10 McArthur GA, Puzanov I, Amaravadi R, Ribas A, Chapman P, Kim KB, Sosman JA, Lee RJ, Nolop K, Flaherty KT, Callahan J, Hicks RJ. Risposte marcate, omogenee e precoci alla tomografia a emissione di positroni [18F]fluorodesossiglucosio a vemurafenib nel melanoma avanzato BRAF-mutante. J Clin Oncol. 2012; 30: 1628-34. https://doi.org/10.1200/JCO.2011.39.1938.
11 Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, Frederick DT, Hurley AD, Nellore A, Kung AL, Wargo JA, Song JS, Fisher DE, et al. BRAF oncogenico regola il metabolismo ossidativo attraverso PGC1alpha e MITF. Cancer Cell. 2013; 23: 302-15. https://doi.org/10.1016/j.ccr.2013.02.003.
12 Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, et al. Miglioramento della sopravvivenza con vemurafenib nel melanoma con mutazione BRAF V600E. N Engl J Med. 2011; 364: 2507-16. https://doi.org/10.1056/NEJMoa1103782.
13 Corazao-Rozas P, Guerreschi P, Jendoubi M, Andre F, Jonneaux A, Scalbert C, Garcon G, Malet-Martino M, Balayssac S, Rocchi S, Savina A, Formstecher P, Mortier L, et al. Lo stress ossidativo mitocondriale è il tallone d’Achille delle cellule di melanoma resistenti all’inibitore Braf-mutante. Oncotarget. 2013; 4: 1986-98. https://doi.org/10.18632/oncotarget.1420.
14 Zhang G, Frederick DT, Wu L, Wei Z, Krepler C, Srinivasan S, Chae YC, Xu X, Choi H, Dimwamwa E, Ope O, Shannan B, Basu D, et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J Clin Invest. 2016; 126: 1834-56. https://doi.org/10.1172/JCI82661.
15 Livingstone E, Swann S, Lilla C, Schadendorf D, Roesch A. Combinare l’inibizione di BRAF(V) (600E) con modulatori del metabolismo bioenergetico mitocondriale per superare la resistenza ai farmaci nel melanoma metastatico. Exp Dermatol. 2015; 24: 709-10. https://doi.org/10.1111/exd.12718.
16 Hoon DS, Spugnardi M, Kuo C, Huang SK, Morton DL, Taback B. Profilazione dell’inattivazione epigenetica dei geni soppressori tumorali nei tumori e nel plasma di pazienti affetti da melanoma cutaneo. Oncogene. 2004; 23: 4014-22. https://doi.org/10.1038/sj.onc.1207505.
17 Dahl C, Christensen C, Jonsson G, Lorentzen A, Skjodt ML, Borg A, Pawelec G, Guldberg P. Mutual exclusivity analysis of genetic and epigenetic drivers in melanoma identifies a link between p14 ARF and RARbeta signaling. Mol Cancer Res. 2013; 11: 1166-78. https://doi.org/10.1158/1541-7786.MCR-13-0006.
18 Lotan R, Lotan D. Enhancement of melanotic expression in cultured mouse melanoma cells by retinoids. J Cell Physiol. 1981; 106: 179-89. https://doi.org/10.1002/jcp.1041060203.
19 Christensen C, Bartkova J, Mistrik M, Hall A, Lange MK, Ralfkiaer U, Bartek J, Guldberg P. Un breve motivo acido in ARF protegge dalla disfunzione mitocondriale e dalla suscettibilità al melanoma. Nat Commun. 2014; 5: 5348. https://doi.org/10.1038/ncomms6348.
20 Baldea I, Costin GE, Shellman Y, Kechris K, Olteanu ED, Filip A, Cosgarea MR, Norris DA, Birlea SA. Effetti bifasici pro-melanogenici e pro-apoptotici dell’acido all-trans-retinoico (ATRA) sui melanociti umani: studio time-course. J Dermatol Sci. 2013; 72: 168-76. https://doi.org/10.1016/j.jdermsci.2013.06.004.
21 Kawakami T, Ohgushi A, Hirobe T, Soma Y. Analisi degli effetti dell’acido all-trans retinoico su melanociti e melanoblasti umani in vitro. J Dermatol. 2017; 44: 93-4. https://doi.org/10.1111/1346-8138.13477.
22 Vazquez F, Lim JH, Chim H, Bhalla K, Girnun G, Pierce K, Clish CB, Granter SR, Widlund HR, Spiegelman BM, Puigserver P. L’espressione di PGC1alpha definisce un sottogruppo di tumori di melanoma umano con una maggiore capacità mitocondriale e resistenza allo stress ossidativo. Cancer Cell. 2013; 23: 287-301. https://doi.org/10.1016/j.ccr.2012.11.020.
23 Donadelli M, Dando I, Fiorini C, Palmieri M. UCP2, una proteina mitocondriale regolata a più livelli. Cell Mol Life Sci. 2014; 71: 1171-90. https://doi.org/10.1007/s00018-013-1407-0.
24 Oberkofler H, Klein K, Felder TK, Krempler F, Patsch W. Ruolo del peroxisome proliferator-activated receptor-gamma coactivator-1alpha nella regolazione trascrizionale del gene umano della proteina di disaccoppiamento 2 nelle cellule INS-1E. Endocrinologia. 2006; 147: 966-76. https://doi.org/10.1210/en.2005-0817.
25 Arigony AL, de Oliveira IM, Machado M, Bordin DL, Bergter L, Pra D, Henriques JA. L’influenza dei micronutrienti nella coltura cellulare: una riflessione sulla vitalità e sulla stabilità genomica. Biomed Res Int. 2013; 2013: 597282. https://doi.org/10.1155/2013/597282.
26 Li Y, Hashimoto Y, Agadir A, Kagechika H, Zhang X. Identificazione di una nuova classe di antagonisti retinoici beta-selettivi del recettore dell’acido retinoico e loro effetti inibitori sull’attività di AP-1 e sull’apoptosi indotta dall’acido retinoico in cellule di cancro al seno umano. J Biol Chem. 1999; 274: 15360-6.
27 de The H, Marchio A, Tiollais P, Dejean A. Espressione differenziale e regolazione dei ligandi dei geni del recettore dell’acido retinoico alfa e beta. EMBO J. 1989; 8: 429-33.
28 Hardie DG, Hawley SA. Proteina chinasi attivata dall’AMP: l’ipotesi della carica energetica rivisitata. Bioessays. 2001; 23: 1112-9. https://doi.org/10.1002/bies.10009.
29 potenzia la loro risposta all’inibizione di BRAFV600E. J Transl Med. 2014; 12: 247. https://doi.org/10.1186/s12967-014-0247-5.
30 Populo H, Caldas R, Lopes JM, Pardal J, Maximo V, Soares P. La sovraespressione della piruvato deidrogenasi chinasi supporta il dicloroacetato come candidato alla terapia del melanoma cutaneo. Expert Opin Ther Targets. 2015; 19: 733-45. https://doi.org/10.1517/14728222.2015.1045416.
31 Kluza J, Corazao-Rozas P, Touil Y, Jendoubi M, Maire C, Guerreschi P, Jonneaux A, Ballot C, Balayssac S, Valable S, Corroyer-Dulmont A, Bernaudin M, Malet-Martino M, et al. L’inattivazione dell’asse di segnalazione HIF-1alpha/PDK3 guida il melanoma verso il metabolismo ossidativo mitocondriale e potenzia l’attività terapeutica dei pro-ossidanti. Cancer Res. 2012; 72: 5035-47. https://doi.org/10.1158/0008-5472.CAN-12-0979.
32 Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, van der Burg SH, Verdegaal EM, Cascante M, Shlomi T, Gottlieb E, Peeper DS. Un ruolo chiave per il gatekeeper mitocondriale piruvato deidrogenasi nella senescenza indotta da oncogeni. Nature. 2013; 498: 109-12. https://doi.org/10.1038/nature12154.
33 Murholm M, Isidor MS, Basse AL, Winther S, Sorensen C, Skovgaard-Petersen J, Nielsen MM, Hansen AS, Quistorff B, Hansen JB. L’acido retinoico ha effetti diversi sull’espressione di UCP1 negli adipociti umani e di topo. BMC Cell Biol. 2013; 14: 41. https://doi.org/10.1186/1471-2121-14-41.
34 Jin W, Xu YP, Yang AH, Xing YQ. Induzione e differenziazione in vitro di cellule staminali mesenchimali del cordone ombelicale in cellule simil-neuronali grazie all’acido all-trans retinoico. Int J Ophthalmol. 2015; 8: 250-6. https://doi.org/10.3980/j.issn.2222-3959.2015.02.07.
35 Tourniaire F, Musinovic H, Gouranton E, Astier J, Marcotorchino J, Arreguin A, Bernot D, Palou A, Bonet ML, Ribot J, Landrier JF. L’acido retinoico tutto trans induce la fosforilazione ossidativa e la biogenesi dei mitocondri negli adipociti. J Lipid Res. 2015; 56: 1100-9. https://doi.org/10.1194/jlr.M053652.
36 Tripathy S, Chapman JD, Han CY, Hogarth CA, Arnold SL, Onken J, Kent T, Goodlett DR, Isoherranen N. L’acido all-trans-retinoico migliora la funzione mitocondriale in modelli di fegato umano. Mol Pharmacol. 2016; 89: 560-74. https://doi.org/10.1124/mol.116.103697.
37 Watabe H, Soma Y, Ito M, Kawa Y, Mizoguchi M. L’acido all-trans retinoico induce la differenziazione e l’apoptosi dei precursori dei melanociti murini con induzione del fattore di trascrizione associato alla microftalmia. J Invest Dermatol. 2002; 118: 35-42. https://doi.org/10.1046/j.0022-202x.2001.01614.x.
38 De Preter G, Neveu MA, Danhier P, Brisson L, Payen VL, Porporato PE, Jordan BF, Sonveaux P, Gallez B. Inhibition of the pentose phosphate pathway by dichloroacetate unravels a missing link between aerobic glycolysis and cancer cell proliferation. Oncotarget. 2016; 7: 2910-20. https://doi.org/10.18632/oncotarget.6272.
39 Michelakis ED, Webster L, Mackey JR. Il dicloroacetato (DCA) come potenziale terapia a bersaglio metabolico per il cancro. Br J Cancer. 2008; 99: 989-94. https://doi.org/10.1038/sj.bjc.6604554.
40 Corazao-Rozas P, Guerreschi P, Andre F, Gabert PE, Lancel S, Dekiouk S, Fontaine D, Tardivel M, Savina A, Quesnel B, Mortier L, Marchetti P, Kluza J. Mitochondrial oxidative phosphorylation controls cancer cell’s life and death decisions upon exposure to MAPK inhibitors. Oncotarget. 2016; 7: 39473-85. https://doi.org/10.18632/oncotarget.7790.
41 Bauer D, Werth F, Nguyen HA, Kiecker F, Eberle J. Ruolo critico delle specie reattive dell’ossigeno (ROS) per il potenziamento sinergico dell’apoptosi da parte di vemurafenib e dell’inibitore del canale del potassio TRAM-34 nelle cellule di melanoma. Cell Death Dis. 2017; 8: e2594. https://doi.org/10.1038/cddis.2017.6.
42 Chen MC, Hsu SL, Lin H, Yang TY. Acido retinoico e trattamento del cancro. Biomedicina (Taipei). 2014; 4: 22. https://doi.org/10.7603/s40681-014-0022-1.
43 Robinson J, Roberts CH, Dodi IA, Madrigal JA, Pawelec G, Wedel L, Marsh SG. Il database europeo delle linee tumorali ricercabili. Cancer Immunol Immunother. 2009; 58: 1501-6. https://doi.org/10.1007/s00262-008-0656-5.
44 Phillips NR, Sprouse ML, Roby RK. Quantificazione simultanea del numero di copie di DNA mitocondriale e del rapporto di delezione: un saggio multiplex di PCR in tempo reale. Sci Rep. 2014; 4: 3887. https://doi.org/10.1038/srep03887.