Sven de Mey 1, Inès Dufait 1, Heng Jiang 1, Cyril Corbet 2, Hui Wang 1, Melissa Van De Gucht 1, Lisa Kerkhove 1, Ka Lun Law 1, Hugo Vandenplas 3, Thierry Gevaert 1, Olivier Feron 2 e Mark De Ridder 1,*
1 Dipartimento di Radioterapia, Universitair Ziekenhuis Brussel, Vrije Universiteit Brussel, 1090 Bruxelles, Belgio; [email protected] (S.d.M.); [email protected] (I.D.); [email protected] (H.J.); [email protected] (H.W.); [email protected] (M.V.D.G.); [email protected] (L.K.); [email protected] (K.L.L.); [email protected] (T.G.)
2 Polo di Farmacologia e Terapeutica (FATH), Institut de Recherche Expérimentale et Clinique (IREC), UCLouvain, 1200 Bruxelles, Belgio; [email protected] (C.C.); [email protected] (O.F.)
3 Dipartimento di Oncologia Medica, Universitair Ziekenhuis Brussel, Vrije Universiteit Brussel, 1090 Bruxelles, Belgio; [email protected]
Corrispondenza: [email protected]
Ricevuto: 14 settembre 2020
Accettato: 4 dicembre 2020
Pubblicato: 9 dicembre 2020
Abstract
Il metabolismo mitocondriale è un bersaglio interessante per la terapia del cancro. La riprogrammazione delle vie metaboliche può potenzialmente sensibilizzare i tumori con opzioni terapeutiche limitate, come il cancro al seno triplo negativo (TNBC), alla chemioterapia e/o alla radioterapia. Il dicloroacetato (DCA) è un inibitore specifico della piruvato deidrogenasi chinasi (PDK), che porta a una maggiore produzione di specie reattive dell’ossigeno (ROS). I ROS sono le principali molecole effettrici delle radiazioni e un loro aumento aumenta la risposta alla radio. In questo studio abbiamo valutato gli effetti del DCA e della radioterapia su due linee cellulari TNBC, EMT6 e 4T1, in condizioni aerobiche e ipossiche. Come previsto, il trattamento con DCA ha ridotto la piruvato deidrogenasi fosforilata (PDH) e ha abbassato il tasso di acidificazione extracellulare (ECAR) e la produzione di lattato. In particolare, il trattamento con DCA ha portato a un aumento significativo della produzione di ROS (fino a 15 volte) nelle cellule tumorali ipossiche, ma non in quelle aerobiche. Coerentemente, il DCA ha radiosensibilizzato le cellule tumorali ipossiche e gli sferoidi 3D, lasciando invariata la radiosensibilità intrinseca delle cellule tumorali. I nostri risultati suggeriscono che, sebbene descritto come un farmaco che promuove la fosforilazione ossidativa (OXPHOS), il DCA può anche aumentare la radioresponsività ipossica. Questo studio apre quindi la strada alla possibilità di intervenire sul metabolismo mitocondriale delle cellule tumorali ipossiche, in particolare per combattere la radioresistenza.
Parole chiave: dicloroacetato; radiosensibilità ipossica; cancro al seno; specie reattive dell’ossigeno
© 2020 dagli autori. Licenziatario MDPI, Basilea, Svizzera. Questo articolo è un articolo ad accesso libero distribuito secondo i termini e le condizioni della licenza Creative Commons Attribution (CC BY) (http://creativecommons.org/licenses/by/4.0/).
INTRODUZIONE
Il cancro al seno è il tumore più comune nelle donne a livello globale e provoca ogni anno 627.000 decessi [1]. Negli ultimi decenni sono stati compiuti progressi significativi nel trattamento del cancro al seno. Tuttavia, sono disponibili solo terapie limitate per le pazienti con tumori al seno triplo-negativi/basali-like [2,3,4]. Lo standard di cura per il trattamento dei tumori al seno ad alto rischio consiste nella chemioterapia neoadiuvante e nella chirurgia, seguite dall’irradiazione post-operatoria di tutto il seno e della parete toracica. Oggi i ricercatori si concentrano sull’ipofrazionamento della radioterapia adiuvante (studio FAST-Forward [5]) o sulla combinazione di chemioterapia e radioterapia preoperatoria. L’approccio radioterapico preoperatorio potrebbe migliorare la sopravvivenza libera da malattia e la qualità di vita [6,7,8,9,10,11].
L’effetto principale delle radiazioni, in particolare di quelle a basso trasferimento di energia lineare, è l’induzione di specie reattive dell’ossigeno (ROS). Durante la radioterapia, i ROS vengono creati dalla radiolisi dell’acqua negli ambienti extracellulari e sono tossici per le cellule tumorali e per i tessuti normali vicini. Circa due terzi dei danni al DNA indotti dalle radiazioni sono attribuiti ai ROS nelle cellule di mammifero [12]. La risposta delle cellule ai danni al DNA indotti dalle radiazioni dipende fortemente dalla presenza di ossigeno. Le molecole di ossigeno possono infatti fissare i danni al DNA prodotti dai radicali liberi. Questa è la cosiddetta “ipotesi della fissazione dell’ossigeno” [12,13]. In assenza di ossigeno, i radicali del DNA vengono ridotti da composti contenenti gruppi sulfidrilici, che riparano il DNA nella sua forma originale. Secondo questa ipotesi, l’ipossia, definita da bassi livelli di ossigeno nel tumore, è una delle principali cause di fallimento clinico della radioterapia [14,15]. L’ipossia è una caratteristica comune del microambiente tumorale. I ROS e l’ipossia sono due fattori con effetti opposti sulla risposta radioterapica del tumore [16]. L’ipotesi generalmente accettata affermava che nelle regioni ipossiche del tumore si verificava un minore stress ossidativo a causa della carenza di ossigeno substrato dei ROS. Tuttavia, recenti evidenze hanno rivelato che in condizioni di ipossia le cellule generano più ROS, soprattutto attraverso il metabolismo mitocondriale [17,18,19,20].
Un tratto distintivo delle cellule tumorali è la capacità di alterare il proprio metabolismo, fornendo l’energia e i metaboliti necessari per la crescita e la sopravvivenza in condizioni di limitazione dei nutrienti e dell’ossigeno. Tuttavia, in presenza di O2, le cellule tumorali adattano il loro metabolismo alla glicolisi, deviando l’ossidazione mitocondriale del piruvato verso la produzione di lattato [21,22]. Questo effetto viene definito effetto Warburg. Rapporti recenti indicano che l’effetto Warburg è implicato nella resistenza allo stress citotossico indotto dalla chemioterapia o dalla radioterapia [23,24,25,26,27]. In questo modo, i metodi di trattamento che bloccano o riducono il metabolismo glicolitico possono aumentare la sensibilità delle cellule tumorali alla radioterapia.
In condizioni di ipossia, l’hypoxia-inducible factor 1-alpha (HIF1α) causa un aumento dell’espressione delle piruvato deidrogenasi chinasi (PDK1-4) [28]. Questi enzimi sono responsabili della commutazione del metabolismo nei mitocondri regolando lo stato di fosforilazione (cioè lo stato di attività) della piruvato deidrogenasi (PDH), che è una delle principali proteine gatekeeper tra la glicolisi e la fosforilazione ossidativa mitocondriale (OXPHOS). Il dicloroacetato (DCA), una piccola molecola inibitrice della PDK, può invertire l’effetto Warburg attivando la PDH e reindirizzando il metabolismo del piruvato nei mitocondri. L’inibizione della PDK da parte del DCA è utilizzata per trattare l’acidosi lattica e le malattie mitocondriali ereditarie [29,30]. Nel complesso, queste osservazioni hanno portato a considerare il DCA come un potenziale farmaco antitumorale [30,31].
È stato dimostrato che il DCA aumenta la radiosensibilità delle cellule tumorali del colon e della prostata, nonché del carcinoma esofageo a cellule squamose e del glioblastoma [32,33,34,35]. Tuttavia, non sono state condotte ricerche simili su cellule di cancro al seno. Il principale meccanismo di radiosensibilizzazione in questi modelli è stato attribuito allo stress ossidativo. Allo stesso tempo, anche l’arresto del ciclo cellulare nella fase G2-M e una ridotta capacità di riserva mitocondriale hanno contribuito agli effetti radiosensibilizzanti. Sulla base dei risultati precedentemente descritti, sono attualmente in corso due studi clinici (uno nel carcinoma della testa e del collo e uno nel glioblastoma) che studiano gli effetti antitumorali del DCA combinato con il trattamento radioterapico [36,37]. Tutte le ricerche precliniche sono state condotte in condizioni aerobiche, ma non sono disponibili dati sugli effetti del trattamento con DCA in condizioni di ipossia. Pertanto, nel presente studio, abbiamo innanzitutto esaminato l’ipotesi che il DCA abbassi il lattato e commuti il metabolismo da un fenotipo glicolitico all’OXPHOS. Successivamente, abbiamo determinato se il DCA può radiosensibilizzare le cellule di cancro al seno ipossiche e abbiamo esaminato ulteriormente i meccanismi sottostanti. I risultati di questo studio possono avere importanti implicazioni per gli studi clinici volti a utilizzare gli inibitori della PDK per migliorare la risposta alla radioterapia nelle pazienti affette da cancro al seno.
Risultati
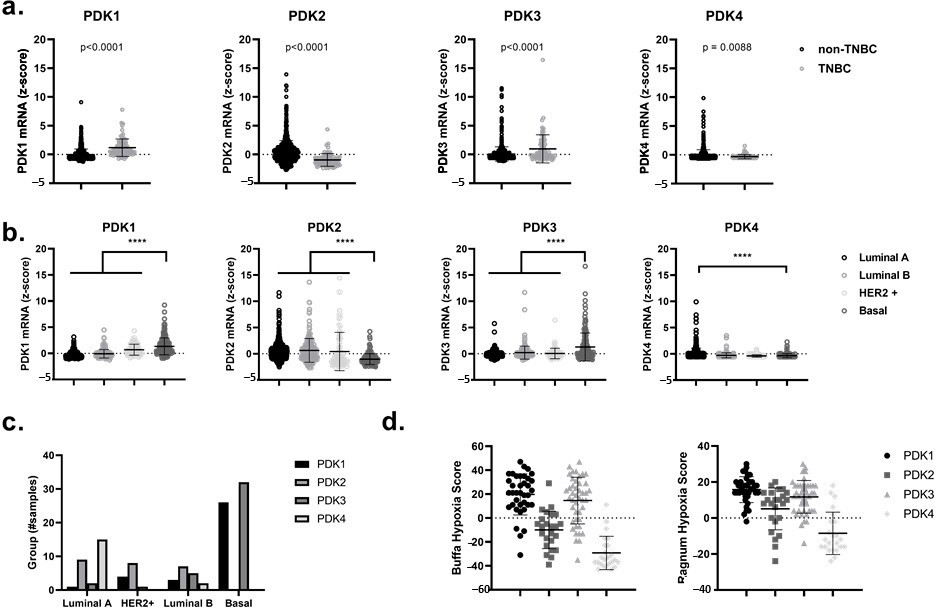
L’elevata espressione di PDK1 e PDK3 nelle pazienti con carcinoma mammario triplo-negativo (TNBC) e basale (Basal-Like Breast Cancer) è correlata a una firma genica correlata all’ipossia
In primo luogo, utilizzando il portale online e pubblicamente accessibile cBioPortal for Cancer Genomics, abbiamo analizzato i livelli di mRNA dei quattro diversi isomeri di PDK, ovvero PDK1, PDK2, PDK3 e PDK4, in dati derivati da pazienti provenienti dal dataset di tumori primari del cancro al seno TCGA (PanCancer Atlas and Cell 2015) [38,39]. Abbiamo evidenziato un aumento significativo (p < 0,0001) dell’espressione di PDK1 e PDK3 nei TNBC rispetto ai non-TNBC e nei tumori al seno di tipo basale rispetto ad altri sottotipi come i tumori al seno luminali A, luminali B e arricchiti di HER2 (Figura 1a-c). In particolare, l’upregulation di PDK1 e PDK3 nelle pazienti con carcinoma mammario potrebbe essere correlata a punteggi di ipossia Ragnum e Buffa più elevati [40,41] (Figura 1d). Questi punteggi di ipossia si basano sull’espressione differenziale di specifici geni correlati all’ipossia e sono liberamente accessibili attraverso il cBioPortal for Cancer Genomics. L’osservata upregulation di PDK1 e PDK3 nei tumori al seno simili al basale e la loro correlazione con un fenotipo ipossico (che è legato all’attivazione di un programma trascrizionale HIF1α-dipendente), suggerisce che l’uso di inibitori di PDK potrebbe servire come un’interessante modalità terapeutica per la radiosensibilizzazione ipossica [42,43,44].
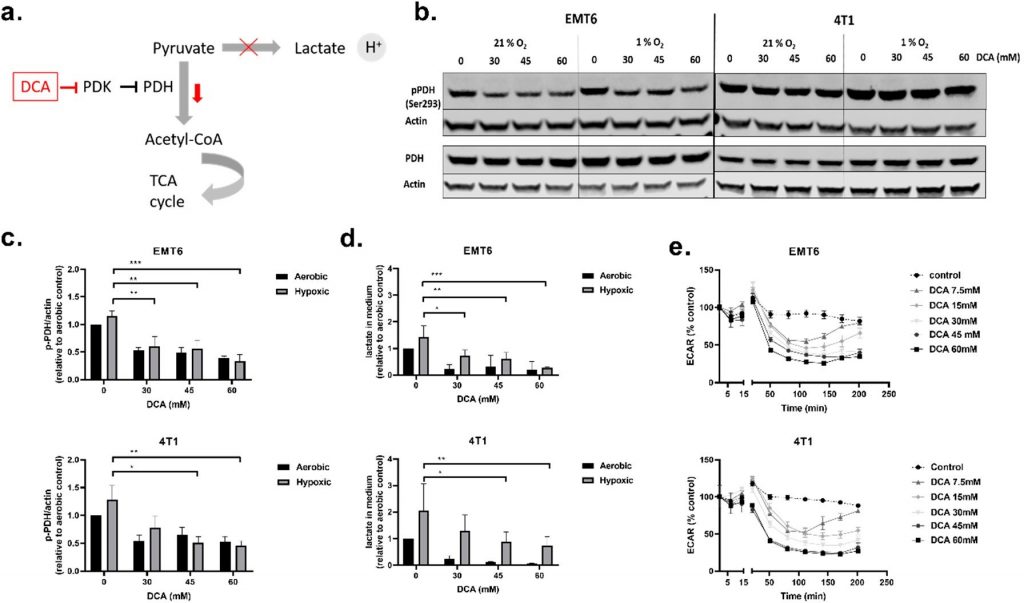
IlDCA ha ridotto la PDH fosforilata, i livelli di lattato extracellulare e il tasso di acidificazione extracellulare (ECAR)
Abbiamo iniziato i nostri esperimenti in vitro eseguendo saggi di vitalità (Figura S1a,b) per determinare le proprietà di inibizione della crescita del DCA. Il DCA ha ridotto la vitalità cellulare in modo dose-dipendente nelle cellule tumorali EMT6 e 4T1, indipendentemente dallo stato di O2 (Figura S1a). Un saggio di proliferazione ha inoltre mostrato che aumentando le concentrazioni di DCA si è osservato un passaggio da un ritardo nella crescita a effetti citostatici e persino citotossici (Figura S1b).
Successivamente, abbiamo analizzato l’influenza del DCA sull’attività di PDK1-4 e sul metabolismo delle cellule TNBC. La PDH è una proteina chiave della glicolisi e dell’OXPHOS mitocondriale, ovvero catalizza la decarbossilazione del piruvato in acetil-CoA. La PDH è inibita attraverso la fosforilazione (su Ser293) da parte della PDK, e questa inibizione può essere invertita attraverso la de-fosforilazione da parte della piruvato deidrogenasi fosfatasi (PDP) [45,46] (Figura 2a). A dosi di tossicità accettabili (30 mM, 45 mM e 60 mM), abbiamo valutato l’effetto del DCA sull’attività della PDK misurando i livelli di PDH fosforilata (p-PDH), il lattato nel mezzo extracellulare e l’ECAR delle cellule in tempo reale (Figura 2b-e). Tutte e tre le dosi di DCA hanno ridotto la quantità di p-PDH nelle cellule EMT6 e 4T1 in modo dose-dipendente sia in condizioni ossigenate che ipossiche. La diminuzione della p-PDH è stata significativa per tutte le dosi di DCA nelle EMT6, mentre nelle 4T1 solo 45 mM e 60 mM hanno ridotto significativamente la p-PDH in condizioni di ipossia (Figura 2b,c). Valutando l’effetto di dosi inferiori di DCA, abbiamo riscontrato che nelle cellule EMT6 la quantità di p-PDH inizia a diminuire quando vengono trattate con 3 mM DCA e nelle cellule 4T1 quando vengono trattate con 10 mM DCA (Figura S2a,b). La quantità di lattato nel mezzo è risultata elevata in condizioni ipossiche rispetto a quelle aerobiche in entrambe le linee cellulari, a sostegno di un aumento netto del turnover glicolitico nelle cellule prive di O2 [47,48]. In accordo con i risultati del Western blot, il trattamento con DCA ha portato a una diminuzione dose-dipendente del lattato nel mezzo per entrambe le linee cellulari (Figura 2d). Sebbene la riduzione del rilascio di lattato sia stata drastica in condizioni aerobiche, una riduzione dose-dipendente della produzione del prodotto glicolitico finale è stata osservata anche in ipossia. Infine, il DCA, a partire da una dose di 7,5 mM, ha causato una riduzione tempo-dipendente dell’ECAR sia nell’EMT6 che nel 4T1 (Figura 2e). Il calo iniziale dell’ECAR dopo il trattamento con dosi inferiori a 30 mM di DCA è stato compensato dopo 2,5 ore in EMT6. In 4T1, abbiamo osservato lo stesso effetto con dosi di DCA inferiori a 15 mM. Questi risultati indicano che il trattamento di linee cellulari TNBC murine con DCA inibisce la fosforilazione di PDH e riduce l’entità della glicolisi sia in condizioni aerobiche che ipossiche.
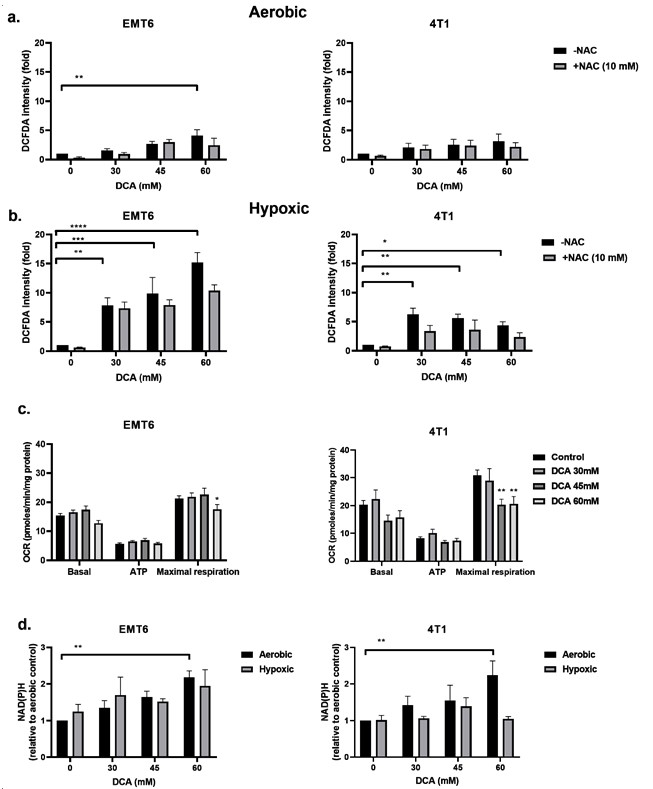
IlDCA induce la produzione di ROS nelle cellule tumorali ipossiche
L’inibizione della PDK e l’attivazione della PDH sono associate all’aumento dei ROS intracellulari [49,50]. I ROS hanno un’importanza fondamentale nel generare danni al DNA in seguito a radiazioni. Abbiamo esaminato i livelli di ROS nelle cellule EMT6 e 4T1 in condizioni aerobiche e ipossiche utilizzando la sonda CM-H2DCFDA. Come mostrato nella Figura 3a,b, il DCA ha innescato una produzione di ROS dose-dipendente sia nelle EMT6 che nelle 4T1. In condizioni aerobiche, solo la dose più alta (60 mM) di DCA ha generato un aumento significativo dei ROS fino a cinque volte rispetto al controllo per le cellule EMT6. Nelle cellule 4T1, in condizioni aerobiche, non è stata rilevata alcuna significativa upregulation dei ROS. L’aumento dei ROS è stato parzialmente contrastato dall’aggiunta dello scavenger dei ROS N-acetilcisteina (NAC). In condizioni di ipossia, abbiamo dimostrato un aumento dose-dipendente dei ROS fino a 15 volte nelle cellule EMT6 e fino a 5 volte nelle cellule 4T1; la dose più bassa di DCA ha effettivamente portato a un aumento significativo dei ROS in entrambi i tipi di cellule (Figura 3b).
Abbiamo pensato che l’aumento della produzione di ROS in condizioni di ipossia potesse derivare dagli effetti combinati dell’alterazione della catena di trasporto degli elettroni mitocondriali (dovuta alla riduzione dell’O2 come accettore finale di elettroni) e del metabolismo ossidativo forzato del piruvato guidato dal DCA. Utilizzando l’analizzatore Seahorse, abbiamo scoperto che il DCA non ha avuto alcun impatto sulla respirazione basale e sulla produzione di ATP, ma ha ridotto significativamente la capacità respiratoria massima nelle cellule tumorali EMT6 e 4T1 (Figura 3c). Un’altra possibilità è che il DCA aumenti i livelli di NAD(P)H, il che è legato a una maggiore produzione di ROS. Abbiamo visto che in condizioni aerobiche, il trattamento con DCA ha avuto un aumento dose-dipendente di NAD(P)H nelle cellule EMT6 e 4T1 (Figura 3d). In condizioni di ipossia, abbiamo dimostrato un possibile aumento di NAD(P)H nelle cellule EMT6 trattate con 60 mM di DCA, ma nessun aumento nelle cellule 4T1 (Figura 3d). Insieme ai dati sui ROS di cui sopra, questi risultati suggeriscono che, sebbene il metabolismo mitocondriale sia ancora preservato, un aumento locale della produzione di ROS in seguito all’esposizione al DCA può alterare l’integrità e la funzione dei mitocondri.
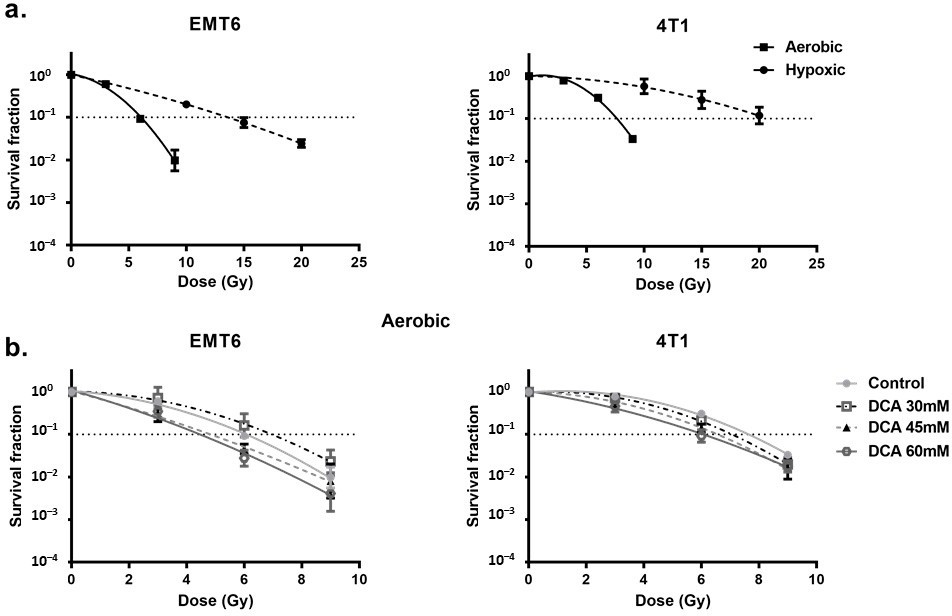
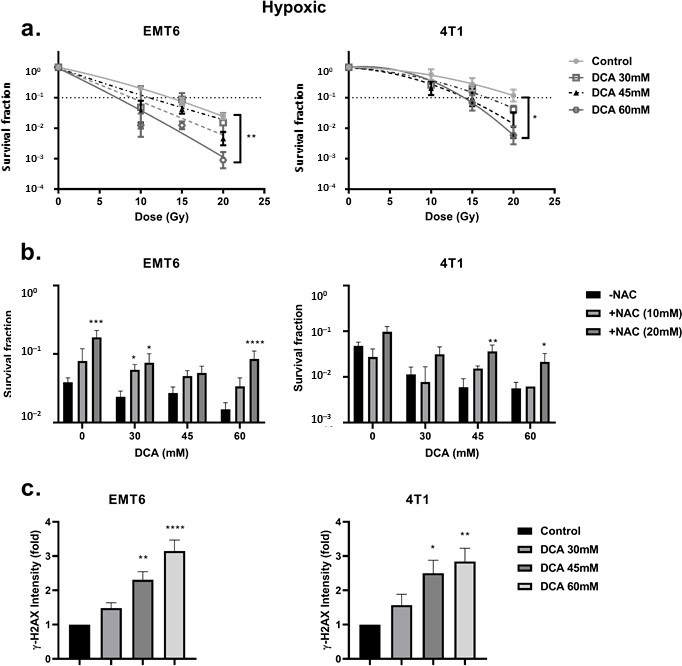
Laradiosensibilità del DCA nelle cellule ipossiche è mediata dai ROS
In primo luogo, abbiamo esaminato la radioresistenza delle cellule indotta dall’ipossia. Abbiamo riscontrato una radioresistenza gravemente compromessa confrontando le condizioni ipossiche con quelle aerobiche, con un rapporto di potenziamento dell’ossigeno di 2,7 e 2,3 per le cellule tumorali EMT6 e 4T1, rispettivamente (Figura 4a). In questo contesto, abbiamo osservato che il trattamento con DCA ha causato un piccolo effetto di radiosensibilizzazione intrinseca nelle cellule EMT6 ma non in quelle 4T1 (Figura 4b). È interessante notare che, in linea con i risultati della generazione di ROS in condizioni di ipossia, il DCA 60 mM ha superato in modo significativo (p < 0,05) la radioresistenza ipossica con rapporti di potenziamento di 2,3 e 1,5 a 60 mM per le cellule tumorali EMT6 e 4T1, rispettivamente (Figura 5a). L’effetto radiosensibilizzante è stato annullato dalla NAC sia nelle cellule EMT6 che in quelle 4T1 (Figura 5b). La causa principale della morte cellulare indotta dalle radiazioni dai ROS è l’induzione di rotture a doppio filamento nel DNA (ds-DNA) [12,51]. Abbiamo quindi esaminato il danno al ds-DNA dopo il trattamento con DCA quantificando lo stato di fosforilazione di γH2AX in condizioni di ipossia. Come mostrato nella Figura 5c, il DCA ha aumentato la formazione di danni al ds-DNA sia nell’EMT6 che nel 4T1 in modo dose-dipendente.
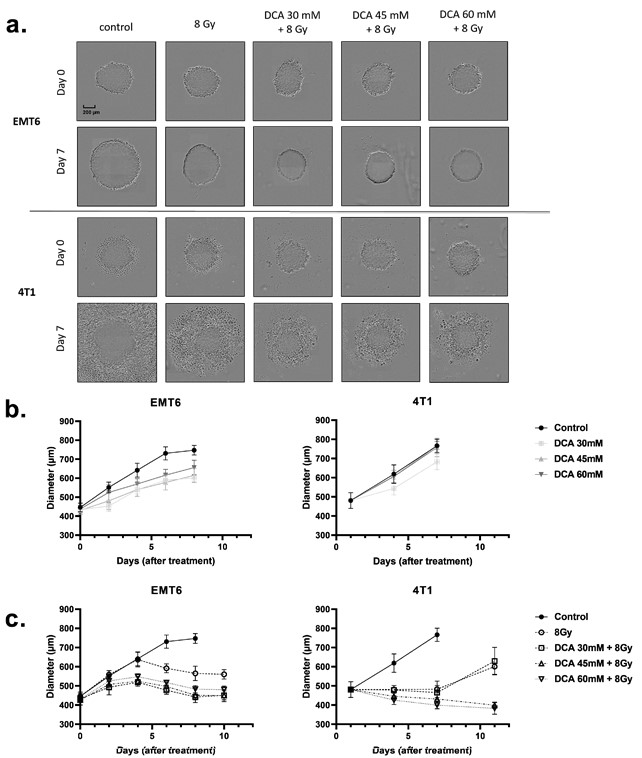
IlDCA radiosensibilizza le colture cellulari 3D (sferoidi)
I risultati di cui sopra ci hanno portato a verificare se il DCA potesse anche migliorare la radiosensibilità di modelli di colture cellulari tridimensionali (3D) (Figura 6a-c) che imitano meglio le proprietà fisico-chimiche del microambiente tumorale, compresi i gradienti di ossigeno. Utilizzando sferoidi ottenuti da colture cellulari EMT6 e 4T1, abbiamo misurato la crescita degli sferoidi dopo il trattamento con DCA e la radioterapia (Figura 6a). Il trattamento con DCA da solo ha provocato un piccolo ritardo nella crescita delle cellule EMT6, ma non ha alterato la crescita degli sferoidi 4T1 (Figura 6b). Un’irradiazione di 8 Gy ha ridotto la crescita degli sferoidi tumorali, un effetto ulteriormente accentuato dalla combinazione con il trattamento con DCA (Figura 6a,c). Da notare che, mentre gli effetti citotossici sono stati osservati negli sferoidi 4T1 (come rivelato dagli aloni di cellule morte), gli effetti citostatici sono stati osservati negli sferoidi EMT6 (Figura 6a).
La combinazione di DCA e radioterapia non ritarda la crescita tumorale in vivo
Abbiamo poi esaminato se il beneficio in vitro della combinazione di DCA e radioterapia potesse essere convalidato in vivo (Figura S3a-d). Topi iniettati con cellule di carcinoma mammario EMT6 o 4T1 sono stati esposti a radiazioni singole (12 Gy o 15 Gy, rispettivamente) (Figura S3a,c) o frazionate (5*4 Gy o 5*6 Gy, rispettivamente) (Figura S3b,d). Le dosi di radiazioni singole e frazionate per tipo di tumore sono simili in termini di dose biologica efficace (BED) e differiscono in funzione della radiosensibilità intrinseca delle linee cellulari utilizzate. È importante notare che l’iniezione di DCA sia i.p. che i.t. per 10 giorni è stata sicura senza indurre una tossicità evidente (Figura S4a-d). L’irradiazione da sola ha ritardato la crescita tumorale in EMT6 per sette giorni con un singolo frazionamento e per quattro giorni con l’irradiazione frazionata (Figura S3a,b). Nei tumori 4T1, le radiazioni hanno ritardato la crescita del tumore per cinque giorni con un singolo frazionamento e per 10 giorni con un frazionamento, come previsto (Figura S3c,d). Il DCA (300 mg/kg), iniettato per via intraperitoneale (ip) o intratumorale (it), non ha ritardato la crescita tumorale e nemmeno la combinazione di DCA e radiazioni (Figura S3a-d). Abbiamo poi verificato se il trattamento con DCA potesse indurre ipossia nei tumori (Figura S3e,f). Sebbene l’entità dell’ipossia evidenziata dal pimonidazolo non sia stata alterata nei tumori EMT6, è stata osservata una tendenza alla diminuzione dell’ipossia in risposta al DCA nei tumori 4T1.
Discussione
Lo scopo di questo studio è stato quello di esaminare se il DCA, mirando al metabolismo mitocondriale, potesse sensibilizzare le cellule di tumore mammario TNBC/basal-like alla radioterapia. La maggior parte dei tumori al seno TNBC e basal-like sono tumori aggressivi per i quali le opzioni terapeutiche sono limitate e la prognosi è infausta [2,3,4]. Questi tumori presentano un fenotipo glicolitico potenziato che supporta la loro prognosi sfavorevole ed è inoltre correlato alla radioresistenza [52]. Nello studio attuale, abbiamo riscontrato che i livelli di mRNA di due delle quattro isoforme PDK (PDK1 e PDK3) sono upregolati nei sottotipi di tumore al seno TNBC e basal-like. La sovraespressione delle PDK è stata rilevata in numerosi campioni di tumori umani [42,53,54,55,56,57,58,59,60,61] e molte linee cellulari tumorali presentano una sostanziale upregolazione delle isoforme PDK [50,62,63]. È stato riportato che la sovraespressione di PDK è associata a una prognosi sfavorevole in diversi tipi di tumore [53,54,55,56,57,58,59,60,61]. La sovraespressione delle PDK nelle cellule tumorali è influenzata da diversi fattori di trascrizione, come HIF1 [28,64]. HIF1 sopprime attivamente l’OXPHOS transattivando direttamente i geni che codificano PDK1 e PDK3. Le PDK a loro volta fosforilano e inattivano la PDH [42]. Pertanto, l’upregulation delle PDK nel cancro può essere ricondotta immediatamente sia alle mutazioni trasformanti sia al microambiente tumorale ipossico. Coerentemente con questi risultati, abbiamo osservato che l’upregulation dell’mRNA di PDK1 e PDK3 è correlata a profili genici legati all’ipossia. La riprogrammazione metabolica, con il bersaglio degli enzimi PDK, per passare dalla glicolisi all’OXPHOS, appare quindi come una promettente via terapeutica per il trattamento dei tumori al seno con opzioni terapeutiche limitate.
Il risultato principale del nostro studio è che l’inibitore PDK DCA può ridurre l’attività glicolitica delle cellule di cancro al seno in presenza di ossigeno ma anche in condizioni di ipossia [29]. Abbiamo scoperto che il DCA abbassa la quantità di PDH fosforilata e innesca una diminuzione dose-dipendente dei livelli di lattato extracellulare e di ECAR nelle cellule aerobiche e ipossiche. Abbiamo quindi combinato DCA e radioterapia, ipotizzando che, invertendo il fenotipo glicolitico e indirizzando più piruvato verso l’ossidazione mitocondriale, le cellule tumorali potessero produrre più ROS e diventare più sensibili alle radiazioni. Effetti intrinseci di radiosensibilizzazione del DCA sono stati riportati in cellule di glioblastoma [34,35], di carcinoma polmonare non a piccole cellule (NSCLC) [65,66], di cancro del colon-retto [35], di cancro della prostata [32] e di medulloblastoma radioresistente [67]. I meccanismi proposti sono l’arresto del ciclo cellulare nella fase G2-M, la creazione di ulteriori danni al DNA e la morte cellulare consecutiva in risposta all’aumento della produzione di ROS mitocondriali. Nello studio attuale, abbiamo scoperto che, mentre sono stati osservati effetti radiosensibilizzanti minimi con la più alta concentrazione non tossica di DCA in condizioni aerobiche, il DCA ha fortemente radiosensibilizzato le cellule di cancro al seno ipossiche, sia in 2D che in sferoidi 3D. Sebbene in teoria i ROS siano associati a meccanismi ossidativi, esiste una collaborazione tra ipossia e ROS nei tumori. L’ipossia aumenta la generazione di ROS attraverso il prolungamento della vita dei radicali semi-chinoni; reciprocamente, i ROS aiutano le cellule tumorali ad adattarsi all’ipossia attraverso la stabilizzazione di HIF1-α [16,68]. Tuttavia, gli insulti extracellulari possono rompere questa partnership innescando un’eccessiva produzione di ROS, che compromette la respirazione mitocondriale e quindi diminuisce la frazione ipossica nei tumori [16,69,70]. In questo contesto, il triossido di arsenico inibisce il consumo di ossigeno delle cellule tumorali attraverso un aumento dei ROS intracellulari, con conseguente aumento della risposta radioelettrica [71]. Anche la soppressione della glicolisi aumenta la risposta radioattiva. Ciò può avvenire tramite ritonavir (inibitore del trasportatore di glucosio), 2-deossiglucosio (inibitore dell’esochinasi) e lonidamina (inibitore dell’esochinasi), che sono in fase di studio clinico in diversi tipi di cancro [72,73,74,75]. Un’altra possibilità è che dopo il trattamento con DCA si creino ROS a causa dell’induzione della NADPH ossidasi [76]. Tuttavia, non sono state trovate prove dirette per concludere che il DCA possa upregolare la NADPH ossidasi [77]. Abbiamo osservato un aumento dose-dipendente di NAD(P)H in condizioni aerobiche, ma non in condizioni di ipossia nelle cellule EMT6 e 4T1. Pertanto, ipotizziamo che l’aumento di NAD(P)H e l’upregolazione delle NADPH ossidasi svolgano solo un ruolo minore nell’aumento dei ROS in condizioni di ipossia. A nostro avviso, il meccanismo primario degli effetti di radiosensibilizzazione osservati deriva molto probabilmente dall’aumento di molte volte della produzione di ROS (fino a 15 volte) dopo il trattamento con DCA in condizioni di ipossia.
In linea con la letteratura, abbiamo dimostrato la necessità di concentrazioni sovrafisiologiche di DCA per suscitare alterazioni dell’attività metabolica, aumento della formazione di ROS e radiosensibilizzazione [29]. La nostra ipotesi centrale è che questi effetti siano la conseguenza dell’inibizione della PDK prodotta dal DCA [78]. Tuttavia, le concentrazioni necessarie per indurre i risultati misurati sono diverse volte superiori alla costante di inibizione (Ki) della PDK1-4. È da notare che il DCA esiste fisiologicamente come anione, è relativamente impermeabile alla membrana nonostante le sue piccole dimensioni e quindi richiede il trasportatore mitocondriale del piruvato per l’assorbimento mitocondriale [79,80]. Tuttavia, la coniugazione del DCA con un trasportatore lipofilo ha migliorato il trasporto mitocondriale. Il valore di IC50 del DCA è stato ridotto da millimolare a micromolare, ben all’interno dell’intervallo Ki della PDK1-4 [81]. Il DCA imita l’efficace inibizione di PDK1-4 da parte del siRNA e il DCA aggiunto al PDK siRNA non ha avuto effetti aggiuntivi [49,50,60,82,83,84,85,86,87,88]. Inoltre, una piccola molecola simile al DCA potrebbe influenzare direttamente o indirettamente altri bersagli cellulari e molecolari. Una recente ricerca ha rilevato che il trattamento con DCA ha aumentato la concentrazione di tutti gli intermedi TCA, ma non ha influenzato l’assorbimento del glucosio o la glicolisi [89]. Altri ricercatori hanno dimostrato che il DCA può aumentare la biosintesi di CoA de novo. Poiché alte concentrazioni di CoA possono essere tossiche per le cellule, questo effetto metabolico potrebbe essere parzialmente responsabile della tossicità delle cellule tumorali mediata dal DCA [90]. Recenti ricerche hanno introdotto una nuova ipotesi, suggerendo che l’efficacia del DCA contro il cancro possa derivare dalla sua capacità di antagonizzare l’acetato. Alti livelli di acetato possono aumentare la sintesi di DNA, RNA e proteine. Inoltre, può essere associato alla resistenza ai farmaci antitumorali [91]. Infine, i ricercatori hanno scoperto che il DCA può attivare la via di segnalazione AMPK, portando a una cascata di effetti metabolici e antitumorali a valle [92,93]. Tuttavia, la nostra ipotesi rimane quella che, in condizioni di ipossia, il passaggio del piruvato nei mitocondri provochi un aumento dei livelli di ROS, con conseguente radiosensibilizzazione delle cellule tumorali. Non siamo riusciti a ricapitolare questi effetti in vivo e sono necessari ulteriori lavori per determinare come tradurre gli effetti radiosensibilizzanti del DCA in compartimenti tumorali ipossici. Si possono escludere problemi di farmacocinetica legati alla somministrazione di DCA in vivo e la dose di DCA utilizzata (150 mg/kg) rientra ampiamente nelle dosi utilizzate in letteratura [29]. La dose umana equivalente alla nostra dose di DCA utilizzata in vivo è di 12 mg/kg/d, ben all’interno della zona di tollerabilità utilizzata negli studi clinici. Una possibile spiegazione del fallimento dei nostri esperimenti in vivo è che è necessaria una dose più elevata di DCA per avere un effetto radiosensibilizzante in vivo. Negli ultimi 30 anni, il DCA è stato somministrato con discreto successo come farmaco sperimentale per il trattamento del diabete di tipo 2, dell’iperlipoproteinemia acquisita e congenita, dell’ischemia miocardica, dell’acidosi lattica acquisita e congenita e, più recentemente, del cancro [29,30]. Diversi studi di fase I/II stanno studiando la sicurezza del DCA e la sua attività come agente antitumorale. Il DCA viene assorbito rapidamente e può persino attraversare la barriera emato-encefalica. Due studi di fase I hanno esaminato la sicurezza del DCA orale in pazienti con tumori cerebrali maligni ricorrenti o metastasi al cervello da tumori del sistema nervoso non centrale [94,95]. Questi studi hanno indicato che il DCA è generalmente ben tollerato dai pazienti. Una spiegazione alternativa per la sconcertante differenza tra gli effetti in vitro e in vivo è in realtà più probabilmente legata ai cambiamenti nei fenotipi metabolici quando le cellule tumorali vengono iniettate (ectopicamente) in vivo. Infatti, l’aumento della radiosensibilità in vitro da parte del DCA si ottiene in condizioni di ipossia e di elevato metabolismo glicolitico, cosicché il passaggio da glicolisi a ossifosfato può essere osservato al momento del trattamento con DCA, insieme a un’ulteriore aumento dei ROS.
L’estensione limitata dell’ipossia nei tumori in vivo può quindi riflettere una capacità limitata del DCA di indurre un cambiamento che è già presente nei tumori ben ossigenati e largamente dipendenti dall’OXPHOS. Sono necessarie ulteriori indagini per verificare gli effetti radiosensibilizzanti del DCA in modelli di tumore mammario di topo caratterizzati da angiogenesi limitata e frazioni ipossiche elevate.
In conclusione, abbiamo dimostrato che il DCA supera la radioresistenza ipossica delle cellule di cancro al seno in sistemi 2D e 3D, che può essere attribuita principalmente all’aumento dei ROS. È notevole che il passaggio dal metabolismo glicolitico a quello ossidativo indotto dal DCA crei anche uno stress ossidativo in condizioni di ipossia. Il DCA è stato utilizzato per molti anni per trattare condizioni metaboliche e malattie mitocondriali ereditarie. Nell’ultimo decennio, il DCA è stato ampiamente riproposto come farmaco antitumorale con dati preclinici promettenti, case report e studi clinici, come descritto in precedenza. Gli attuali risultati preclinici indicano che sono necessari ulteriori approfondimenti sul potenziale antitumorale del DCA, considerando che le cellule tumorali ipossiche non sono risparmiate dall’inibitore PDK che può indurre uno stress ossidativo letale, in particolare se associato alla radioterapia.
Materiali e metodi
Analisi della coorte del cancro al seno TCGA
I profili di espressione dell’mRNA di PDK1-4 (RNA Seq V2 RSEM o log RNA Seq V2 RSEM) sono stati interrogati dal sito web cBioPortal sotto forma di dati trasformati in z-score [38,39]. I dati interrogati sono stati valutati da 1084 casi di cancro al seno pubblicamente disponibili del TCGA PanCancer Atlas e da 817 casi di cancro al seno del database TCGA Cell 2015. Per il set di dati TCGA Cell 2015, l’analisi di PDK1-4 è stata eseguita confrontando la sottopopolazione triplo-negativa con il resto dei casi di cancro al seno. Il cancro al seno triplo negativo è stato determinato da uno stato “negativo” per i punteggi immunoistochimici dei geni del recettore degli estrogeni (ER), del recettore del progesterone (PR) e del recettore 2 del fattore di crescita epidermico umano (HER2) (in totale 83 casi). Nel database TCGA PanCancer Atlas, l’analisi dell’espressione di PDK1-4 è stata eseguita in diverse categorie Pam50. Pam50 è una firma di 50 geni che classifica il cancro al seno in cinque sottotipi molecolari intrinseci: Luminale A, Luminale B, arricchito di HER2, Basale e Normale. L’espressione dell’mRNA del tumore al seno TCGA e i dati clinici sono stati analizzati direttamente sul sito web cBioPortal o scaricati per ulteriori analisi. L’analisi del punteggio di ipossia di Buffa e Ragnum dei campioni in cui PDK1-4 ha un’espressione di z-score superiore a 2 è stata effettuata direttamente sul sito web cBioPortal.
Linee cellulari e sostanze chimiche
La linea cellulare di adenocarcinoma mammario murino EMT6 è stata gentilmente fornita da Edith Lord (University of Rochester, Cancer Center, New York) e le cellule 4T1 sono state ottenute dalla American Type Culture Collection. Tutti gli esperimenti sono stati eseguiti in terreno Roswell Park Memorial Institute 1640 (Thermo Fisher Scientific, Waltham, MA, USA) integrato con il 10% di siero fetale bovino (Greiner Bio-One, Kremsmünster, Austria). Per tutti i trattamenti delle cellule è stato utilizzato il tampone HEPES. Le sostanze chimiche sono state ottenute da Sigma-Aldrich (Sigma-Aldrich, St. Louis, MO, USA), salvo diversa indicazione
Trattamenti
EMT6 e 4T1 sono state fatte crescere fino alla confluenza e trattate con DCA per 16 ore alle concentrazioni indicate. La N-acetilcisteina (NAC) è stata aggiunta a 10 mM o 20 mM alle colture sia 1 ora prima che durante il trattamento con DCA. Successivamente, le colture sono state utilizzate per ulteriori analisi come descritto di seguito. Il trattamento è stato eseguito in condizioni aerobiche o ipossiche. L’ipossia è stata indotta dall’incubazione in un gas bilanciato azoto/diossido di carbonio contenente l’1% di ossigeno [96]
Test MTT
La citotossicità del DCA è stata valutata mediante il test MTT come descritto altrove <a href=”#97″>[97,</a></sup><a href=”#98″><sup>98]</sup>.</a> In breve, le cellule sono state coltivate in piastre da 96 pozzetti e trattate con le concentrazioni indicate. Dopo il trattamento, il terreno è stato aspirato e sono stati aggiunti 50 µL di reagente MTT (5 mg/mL) per 1,5 h. Successivamente, sono stati aggiunti 200 µL di solvente MTT (19:1 DMSO/HCL) e mescolati per dissolvere i cristalli di formazan generati all’interno delle cellule. L’assorbanza è stata misurata alla lunghezza d’onda di 540 nm utilizzando uno spettrofotometro (Bio-Rad Laboratories, Hercules, CA, USA). La vitalità cellulare è stata determinata normalizzando le cellule trattate rispetto a quelle di controllo non trattate.</p>
<p><strong>Saggio di crescita cinetica<br></strong>L’influenza del DCA sulla proliferazione delle cellule EMT6 e 4T1 è stata valutata utilizzando un saggio di crescita cinetica. Le cellule sono state fatte crescere fino alla confluenza in piastre di coltura a 96 pozzetti e trattate con DCA alle concentrazioni indicate in 6 repliche. Le microfotografie sono state scattate ogni due ore utilizzando un imager per cellule vive Incucyte (Essen Biosciences, Newark, Regno Unito) e la confluenza delle colture è stata misurata utilizzando il software Incucyte (Incucyte ZOOM 2018A, Essen Biosciences) su 80 h in coltura.</p>
<p><strong>Western Blot<br></strong>Le analisi Western blot sono state eseguite come precedentemente descritto <sup><a href=”#99″>[99]</a></sup>. In breve, le cellule sono state lisate in un tampone triton-X all’1% integrato con un inibitore della fosfatasi (P5726), un inibitore della proteasi (P8340) e leupeptina trifluoroacetato (L2023). I lisati sono stati centrifugati e la concentrazione proteica è stata determinata con il test delle proteine DC Bio-Rad (Bio-Rad 500-0116). Quantità equivalenti di proteine sono state caricate su un gel di acrilammide risolvente al 12%. Il trasferimento delle proteine è stato condotto per una notte a 4 °C utilizzando una membrana di nitrocellulosa (0,45 µM, Thermo 88018, Thermo Fisher Scientific). Le membrane sono state bloccate con BSA al 5% in TBS e lavate (TBST). Le membrane bloccate sono state marcate con l’anticorpo primario durante la notte a 4 °C. Gli anticorpi primari sono stati marcati con anticorpi secondari nel vicino infrarosso (IRDyes 680 RD o 800 CW, LI-COR Biosciences, Lincoln, NE, USA), rilevati e quantificati con Odyssey Fc Imaging System (LI-COR Biosciences). Gli anticorpi primari erano: fosfo-PDH (ABS204, MERCK, Darmstadt, Germania), PDH totale (C54G1, cell signaling), antibeta ACTIN (A1978) e antialfa TUBULIN (T9026).</p>
<p><strong>Saggio del lattato<br></strong>Dopo il trattamento, è stato eseguito il saggio del L-lattato, secondo le istruzioni del produttore. In breve, il surnatante delle cellule è stato prelevato e introdotto nella miscela di reazione master. Successivamente, è stato effettuato un periodo di incubazione di 30 minuti a temperatura ambiente e al buio. Successivamente, è stata misurata l’assorbanza a 570 nm ed è stata calcolata la quantità di L-lattato nel mezzo. Inoltre, le cellule sono state lisate e la quantità totale di proteine è stata esaminata con il BCA protein assay (23227, Thermo Fisher); questa fase è stata eseguita per normalizzare i valori di lattato alla quantità di proteine nella cellula.</p>
<p><strong>Profilo metabolico Seahorse<br></strong>Il tasso di consumo di ossigeno (OCR) e il tasso di acidificazione extracellulare (ECAR) sono stati determinati utilizzando un analizzatore Seahorse XF96 (Agilent Technologies, Santa Clara, CA, USA) come precedentemente riportato <sup><a href=”#100″>[100]</a></sup>. In breve, 1,5 × 10<sup>5</sup> cellule sono state seminate in piastre da 96 pozzetti. Le cellule sono state poi trattate con DCA, per tutta la notte (per le misurazioni OCR) o per 3 ore (durante la corsa Seahorse, per l’ECAR). Le cellule sono state equilibrate in terreno Dulbecco’s Modified Eagle Medium (DMEM) non tamponato con 2 mM di glutammina e 10 mM di glucosio a 37 °C in un incubatore privo di CO<sub>2</sub> e poi misurate con un analizzatore Seahorse. Per estrarre informazioni dettagliate sulla catena di trasporto degli elettroni nei mitocondri, sono stati aggiunti in sequenza inibitori specifici costituiti da oligomicina, FCCP, rotenone e antimicina A. L’ECAR è stato normalizzato al livello basale.</p>
<p><strong>Produzione di ROS<br></strong>Il livello intracellulare di ROS è stato rilevato utilizzando il 5-(6)-clorometil-2′,7′-diclorodiidro-fluoresceina diacetato (CM-H<sub>2</sub>DCFDA), una sonda fluorescente sensibile all’ossidazione (Abcam, Cambridge, Regno Unito) come precedentemente descritto <sup><a href=”#97″>[97]</a></sup>. In breve, dopo il trattamento, le cellule sono state colorate con 5 μM CM-H2DCFDA a 37 °C per 30 minuti. L’intensità media della fluorescenza è stata misurata da un citometro a flusso FACSCanto (BD Bioscience, Franklin lakes, NJ, USA) e analizzata dal software Flowjo (BD Bioscience).</p>
<p><strong>Misurazione del NAD(P)H<br></strong>Il livello intracellulare di NAD(P)H è stato rilevato utilizzando il kit di analisi citometrica a flusso Cell Meter<sup>TM</sup> intracellulare NADH/NADPH (AAT BioQuest, Sunnyvale, CA, USA), secondo le istruzioni del produttore. In breve, dopo il trattamento, le cellule sono state colorate con il sensore NAD(P)H JZLA707 a 37 °C per 45 minuti. L’intensità media della fluorescenza è stata misurata con un citometro a flusso FACSCanto (BD Bioscience) e analizzata con il software Flowjo (BD Bioscience).</p>
<p><strong>Saggio di irradiazione e clonogenicità<br></strong>Dopo il trattamento, le cellule sono state irradiate alle dosi indicate su un Linac da 6 MV (Varian Truebeam STx, Palo Alto, CA, USA; BrainLAB AG, Feldkirchen, Germania) e riseminate in piastre a 6 pozzetti per la formazione di colonie. Prima della semina, le cellule sono state contate e normalizzate rispetto alle condizioni di controllo. Dopo 7-12 giorni, le colture sono state fissate con cristalvioletto e sono state contate le colonie (>50 cellule). Le frazioni di sopravvivenza (SF) sono state adattate al modello lineare-quadratico utilizzando il software GraphPad Prism 8 (GraphPad Prism Software Inc, San Diego, CA, USA). La radiosensibilizzazione è stata valutata al livello di 0,1 frazioni di sopravvivenza.</p>
<p><strong>Colture cellulari tridimensionali (3D) (sferoidi)<br></strong>Sferoidi sono stati preparati con cellule EMT6 e 4T1 seminando 4000 cellule/pozzetto in una piastra a 96 pozzetti a bassissimo attacco (Corning, Corning, NY, USA). Il DCA è stato aggiunto al terreno di coltura quando gli sferoidi avevano un diametro di circa 500 µm. In seguito, gli sferoidi sono stati irradiati a 8 Gy e il trattamento è stato lavato con terreno fresco, rinfrescato ogni 3 giorni. La crescita degli sferoidi è stata monitorata con il sistema IncuCyte Live Cell Imaging System (Essen Bioscience) per 10 giorni.</p>
<p><strong>Modello tumorale di topo<br></strong> Le cellule tumorali 4T1 e EMT6 (0,5 × 10<sup>6</sup>) sono state inoculate nell’arto posteriore sinistro di topi Balb/c (femmine, 7-9 settimane di età; Charles River Laboratories, L’Arbresle Cedex, Francia). Quando i tumori hanno raggiunto circa 150 mm<sup>3</sup>, i topi sono stati randomizzati e trattati per 10 giorni consecutivi con DCA 300 mg/kg (intraperitoneale o intratumorale). I topi sono stati irradiati con una dose singola di 12 Gy (tumori EMT6) o 15 Gy (tumori 4T1) o con uno schema di irradiazione frazionata di 5*4 Gy (tumori EMT6) e 5*6 Gy (tumori 4T1) quando i tumori hanno raggiunto circa 150 mm3. Le radiazioni sono state erogate con un Linac da 6 MV (Varian Truebeam STx). Durante l’intero corso dell’esperimento, i tumori sono stati misurati con un calibro elettronico e il volume del tumore è stato calcolato con la formula: Volume = (Lunghezza × Larghezza<sup>2</sup>) × 0,5. Gli esperimenti sono stati approvati dal Comitato etico per l’uso di animali da laboratorio della Vrije Universiteit Brussel (dossier etico n: 16-552-2 (18/4/2017) e 18-552-2 (1/6/2018) </p>
<p><strong>Colorazione con pimonidazolo su sezioni tumorali<br></strong>I tumori sono stati inoculati come descritto nella parte 4.13. Dopo il trattamento, il pimonidazolo (60 mg/kg; Hypoxyprobe) è stato iniettato i.v. nella vena della coda. I tumori sono stati escissi 1,5 ore dopo, pesati, congelati a caldo e conservati in fiale di plastica a -80 °C. Le sezioni tumorali (5 µm) sono state quindi sottoposte a immunocolorazione utilizzando un anticorpo di coniglio anti-Pimo (Hypoxyprobe, Burlington, MA, USA), che è stato colorato con un anticorpo FITC anti-coniglio (Abcam). I vetrini tumorali sono stati montati con liquido di montaggio (DAKO mounting medium, Agilent) miscelato con dapi (Sigma-Aldrich) e coperti con un coprioggetto. Le immagini sono state acquisite con microscopia confocale a fluorescenza (EVOS FL, Thermo Fisher) e analizzate con ImageJ.</p>
<p><strong>Statistiche</strong><br>Tutte le analisi sono state eseguite con GraphPad Prism 8.4.3. I dati sono espressi come media ± SEM di almeno tre esperimenti indipendenti, salvo diversa indicazione. Per le analisi statistiche sono stati utilizzati il test <em>t</em> non comparato, l’ANOVA a una via seguita dal test di confronto multiplo di Dunnett e l’ANOVA a due vie con il test di confronto multiplo di Dunnett, Sidak o Tukey: * <em>p</em> < 0,05, ** <em>p</em> < 0,01, *** <em>p</em> < 0,001, **** <em>p</em> < 0,0001.</p>
<h2>Materiale complementare</h2>
<p>I materiali di supporto sono disponibili all’indirizzo https://www.mdpi.com/1422-0067/21/24/9367/s1.</p>
<h2>Contributi dell’autore</h2>
<p>Concettualizzazione, S.d.M., I.D. e H.J.; cura dei dati, S.d.M., C.C., K.L.L. e H.V.; indagine, S.d.M.; metodologia, T.G.; supervisione, I.D., H.J., O.F. e M.D.R.; visualizzazione, S.d.M.; stesura della bozza originale, S.d.M.; revisione ed editing, I.D., C.C., H.W., M.V.D.G., L.K., O.F. e M.D.R. Tutti gli autori hanno discusso i risultati e contribuito alla stesura del manoscritto finale. Tutti gli autori hanno letto e approvato la versione pubblicata del manoscritto.</p>
<h2>Finanziamento</h2>
<p>Questo lavoro è stato sostenuto dal programma strategico di ricerca “Societal Benefit of Markerless Stereotactic Body Radiotherapy: a Statistical Support based on Quantitative Imaging” (Zwaartepunt, SRP 53, 2019-2024) del consiglio di ricerca della Vrije Universiteit Brussel.</p>
<h2>Riconoscimenti</h2>
<p>Gli autori ringraziano Valeri Verovski per i preziosi e costruttivi suggerimenti.</p>
<h2>Conflitti di interesse</h2>
<p>Gli autori dichiarano di non avere potenziali conflitti di interesse.</p>
<h2>Nota dell’editore:</h2>
<p>MDPI rimane neutrale rispetto alle rivendicazioni giurisdizionali nelle mappe pubblicate e alle affiliazioni istituzionali.</p>
<h2>Abbreviazioni</h2>
<figure class=”wp-block-table”><table><tbody><tr><td>DCA</td><td>Dicloroacetato</td></tr><tr><td>ECAR</td><td>Tasso di acidificazione extracellulare</td></tr><tr><td>NAC</td><td>N-acetil-cisteina</td></tr><tr><td>OCR</td><td>Tasso di consumo di ossigeno</td></tr><tr><td>OXPHOS</td><td>Fosforilazione ossidativa</td></tr><tr><td>PDH</td><td>Piruvato deidrogenasi</td></tr><tr><td>PDK</td><td>Piruvato deidrogenasi chinasi</td></tr><tr><td>PDP</td><td>Piruvato deidrogenasi fosfatasi</td></tr><tr><td>ROS</td><td>Specie reattive dell’ossigeno</td></tr><tr><td>TBNC</td><td>Cancro al seno triplo-negativo al seno</td></tr></tbody></table></figure>
<h2>REFERENZE</h2>
<span id=”1″ class=”referencess blue-text”>1</span> Organizzazione Mondiale della Sanità. Cancro al seno. Disponibile online: https://www.who.int/cancer/prevention/ diagnosis-screening/breast-cancer/en/ (consultato il 10 settembre 2020).
<br><span id=”2″ class=”referencess blue-text”>2</span> Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Il cancro al seno triplo negativo: Sfide e opportunità di una malattia eterogenea. Nat. Rev. Clin. Oncol. 2016, 13, 674-690. [CrossRef] [PubMed]
<br><span id=”3″ class=”referencess blue-text”>3</span> Fragomeni, S.M.; Sciallis, A.; Jeruss, J.S. Molecular subtypes and local-regional control of breast cancer. Surg. Oncol. Clin. N. Am. 2018, 27, 95-120. [CrossRef] [PubMed]
<br><span id=”4″ class=”referencess blue-text”>4</span> Prat, A.; Pineda, E.; Adamo, B.; Galván, P.; Fernández, A.; Gaba, L.; Díez, M.; Viladot, M.; Arance, A.; Muñoz, M. Clinical implications of the intrinsic molecular subtypes of breast cancer. Breast 2015, 24 (Suppl. 2), S26-S35. [CrossRef] [PubMed]
<br><span id=”5″ class=”referencess blue-text”>5</span> Brunt, A.M.; Haviland, J.S.; Wheatley, D.A.; Sydenham, M.A.; Alhasso, A.; Bloomfield, D.J.; Chan, C.; Churn, M.; Cleator, S.; Coles, C.E.; et al. Radioterapia ipofrazionata al seno per 1 settimana versus 3 settimane (FAST-Forward): 5-year efficacy and late normal tissue effects results from a multicentre, non-inferiority, randomised, phase 3 trial. Lancet 2020, 395, 1613-1626. [CrossRef]
<br><span id=”6″ class=”referencess blue-text”>6</span> Poleszczuk, J.; Luddy, K.; Chen, L.; Lee, J.K.; Harrison, L.B.; Czerniecki, B.J.; Soliman, H.; Enderling, H. Neoadjuvant radiotherapy of early-stage breast cancer and long-term disease-free survival. Breast Cancer Res. 2017, 19, 1-7. [CrossRef]
<br><span id=”7″ class=”referencess blue-text”>7</span> Lightowlers, S.V.; Boersma, L.J.; Fourquet, A.; Kirova, Y.M.; Offersen, B.V.; Poortmans, P.; Scholten, A.N.; Somaiah, N.; Coles, C.E. Preoperative breast radiation therapy: Indicazioni e prospettive. Eur. J. Cancer 2017, 82, 184-192. [CrossRef]
<br><span id=”8″ class=”referencess blue-text”>8</span> Palta, M.; Yoo, S.; Adamson, J.D.; Prosnitz, L.R.; Horton, J.K. Preoperative single fraction partial breast radiotherapy for early-stage breast cancer. Int. J. Radiat. Oncol. Biol. Phys. 2012, 82, 37-42. [CrossRef]
<br><span id=”9″ class=”referencess blue-text”>9</span> Horton, J.K.; Blitzblau, R.C.; Yoo, S.; Geradts, J.; Chang, Z.; Baker, J.A.; Georgiade, G.S.; Chen, W.; Siamakpour-Reihani, S.; Wang, C.; et al. Preoperative Single-Fraction Partial Breast Radiation Therapy: A Novel Phase 1, Dose-Escalation Protocol with Radiation Response Biomarkers. Int. J. Radiat. Oncol. Biol. Phys. 2015, 92, 846-855. [CrossRef]
<br><span id=”10″ class=”referencess blue-text”>10</span> Roth, S.L.; Audretsch, W.; Bojar, H.; Lang, I.; Willers, R.; Budach, W. Retrospective study of neoadjuvant versus adjuvant radiochemotherapy in locally advanced noninflammatory breast cancer: Vantaggio di sopravvivenza nella categoria cT2 con la radiochemioterapia neoadiuvante. Strahlentherapie und Onkologie 2010, 186, 299-306. [CrossRef]
<br><span id=”11″ class=”referencess blue-text”>11</span> Radioterapia accelerata pre- o post-operatoria. Disponibile online: https://ClinicalTrials.gov/show/ NCT03783364 (consultato il 27 agosto 2020).
<br><span id=”12″ class=”referencess blue-text”>12</span> Hall, E.J.; Giaccia, A.J. Radiobiology for the Radiologist; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2012; Volume 7.
<br><span id=”13″ class=”referencess blue-text”>13</span> De Ridder, M.; Tournel, K.; Van Nieuwenhove, Y.; Engels, B.; Hoorens, A.; Everaert, H.; De Beeck, B.O.; Vinh-Hung, V.; De Grève, J.; Delvaux, G.; et al. Phase II study of preoperative helical tomotherapy for rectal cancer. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 728-734. [CrossRef]
<br><span id=”14″ class=”referencess blue-text”>14</span> Gray, L.H.; Conger, A.D.; Ebert, M.; Hornsey, S.; Scott, O.C. The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. Br. J. Radiol. 1953, 26, 638-648. [CrossRef] [PubMed]
<br><span id=”15″ class=”referencess blue-text”>15</span> Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437-447. [CrossRef] [PubMed]
<br><span id=”16″ class=”referencess blue-text”>16</span> Wang, H.; Jiang, H.; Van De Gucht, M.; De Ridder, M. Hypoxic Radioresistance: I ROS possono essere la chiave per superarla? Cancers 2019, 11, 112. [CrossRef] [PubMed]
<br><span id=”17″ class=”referencess blue-text”>17</span> Guzy, R.D.; Schumacker, P.T. Oxygen sensing by mitochondria at complex III: The paradox of increased reactive oxygen species during hypoxia. Exp. Physiol. 2006, 91, 807-819. [CrossRef]
<br><span id=”18″ class=”referencess blue-text”>18</span> Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275. [CrossRef]
<br><span id=”19″ class=”referencess blue-text”>19</span> Azimi, I.; Petersen, R.M.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Le specie reattive dell’ossigeno indotte dall’ipossia mediano l’espressione di N-caderina e SERPINE1, la segnalazione di EGFR e la motilità nelle cellule di cancro al seno MDA-MB-468. Sci. Rep. 2017, 7, 1-11. [CrossRef]
<br><span id=”20″ class=”referencess blue-text”>20</span> Johnson, M.K.; Vathanayagam, R.R.; Wang, E.S. Hypoxia-Associated Effects on Reactive Oxygen Species Generation by Human Acute Myeloid Leukemia Cells. Blood 2011, 118, 4998. [CrossRef]
<br><span id=”21″ class=”referencess blue-text”>21</span> Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: La prossima generazione. Cell 2011, 144, 646-674. [CrossRef]
<br><span id=”22″ class=”referencess blue-text”>22</span> Warburg, O. Sull’origine delle cellule cancerose. Science 1956, 123, 309-314. [CrossRef]
<br><span id=”23″ class=”referencess blue-text”>23</span> Pitroda, S.P.; Wakim, B.T.; Sood, R.F.; Beveridge, M.G.; Beckett, M.A.; MacDermed, D.M.; Weichselbaum, R.R.; Khodarev, N.N. STAT1-dependent expression of energy metabolic pathways links tumour growth and radioresistance to the Warburg effect. BMC Med. 2009, 7, 68. [CrossRef]
<br><span id=”24″ class=”referencess blue-text”>24</span> Song, K.; Li, M.; Xu, X.; Xuan, L.I.; Huang, G.; Liu, Q. La resistenza alla chemioterapia è associata ad alterazioni del metabolismo del glucosio nella leucemia mieloide acuta. Oncol. Lett. 2016, 12, 334-342. [CrossRef]
<br><span id=”25″ class=”referencess blue-text”>25</span> Zhou, Y.; Tozzi, F.; Chen, J.; Fan, F.; Xia, L.; Wang, J.; Gao, G.; Zhang, A.; Xia, X.; Brasher, H.; et al. Intracellular ATP levels are a pivotal determinant of chemoresistance in colon cancer cells. Cancer Res. 2012, 72, 304-314. [CrossRef] [PubMed]
<br><span id=”26″ class=”referencess blue-text”>26</span> Shimura, T.; Noma, N.; Sano, Y.; Ochiai, Y.; Oikawa, T.; Fukumoto, M.; Kunugita, N. AKT-mediated enhanced aerobic glycolysis causes acquired radioresistance by human tumor cells. Radiother. Oncol. 2014, 112, 302-307. [CrossRef] [PubMed]
<br><span id=”27″ class=”referencess blue-text”>27</span> Sattler, U.G.; Meyer, S.S.; Quennet, V.; Hoerner, C.; Knoerzer, H.; Fabian, C.; Yaromina, A.; Zips, D.; Walenta, S.; Baumann, M.; et al. Glycolytic metabolism and tumour response to fractionated irradiation. Radiother. Oncol. 2010, 94, 102-109. [CrossRef] [PubMed]
<br><span id=”28″ class=”referencess blue-text”>28</span> Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated Expression of Pyruvate Dehydrogenase Kinase: A Metabolic Switch Required for Cellular Adaptation to Hypoxia. Cell Metab. 2006, 3, 177-185. [CrossRef] [PubMed]
<br><span id=”29″ class=”referencess blue-text”>29</span> Kankotia, S.; Stacpoole, P.W. Dichloroacetate and cancer: Una nuova casa per un farmaco orfano? Biochim. Biophys. Acta 2014, 1846, 617-629. [CrossRef]
<br><span id=”30″ class=”referencess blue-text”>30</span> James, M.O.; Jahn, S.C.; Zhong, G.; Smeltz, M.G.; Hu, Z.; Stacpoole, P.W. Therapeutic applications of dichloroacetate and the role of glutathione transferase zeta-1. Pharmacol. Ther. 2017, 170, 166-180. [CrossRef] <br><span id=”31″ class=”referencess blue-text”>31</span> Tataranni, T.; Piccoli, C. Dicloroacetato (DCA) e cancro: Una panoramica sulle applicazioni cliniche. Oxidative Med. Cell. Longev. 2019, 2019, 1-14. [CrossRef]
<br><span id=”32″ class=”referencess blue-text”>32</span> Cao, W.; Yacoub, S.; Shiverick, K.T.; Namiki, K.; Sakai, Y.; Porvasnik, S.; Urbanek, C.; Rosser, C.J. Il dicloroacetato (DCA) sensibilizza in vitro alle radiazioni sia le cellule di cancro alla prostata wild-type che quelle che esprimono Bcl-2. Prostata 2008, 68, 1223-1231. [CrossRef]
<br><span id=”33″ class=”referencess blue-text”>33</span> Dong, G.; Chen, Q.; Jiang, F.; Yu, D.; Mao, Q.; Xia, W.; Shi, R.; Wang, J.; Xu, L. Diisopropylamine Dichloroacetate Enhances Radiosensitization in Esophageal Squamous Cell Carcinoma by Increasing Mitochondria-Derived Reactive Oxygen Species Levels. Oncotarget 2016, 7, 68170-68178. [CrossRef]
<br><span id=”34″ class=”referencess blue-text”>34</span> Shen, H.; Hau, E.; Joshi, S.; Dilda, P.J.; McDonald, K.L. Sensitization of Glioblastoma Cells to Irradiation by Modulating the Glucose Metabolism. Mol. Cancer Ther. 2015, 14, 1794-1804. [CrossRef] [PubMed]
<br><span id=”35″ class=”referencess blue-text”>35</span> Zwicker, F.; Kirsner, A.; Peschke, P.; Roeder, F.; Debus, J.; Huber, P.E.; Weber, K.J. Dichloroacetate induce una radiosensibilità tumore-specifica in vitro ma attenua il ritardo di crescita tumorale indotto dalle radiazioni in vivo. Strahlentherapie und Onkologie 2013, 189, 684-692. [CrossRef] [PubMed]
<br><span id=”36″ class=”referencess blue-text”>36</span> Combinazione di radioterapia e temozolomide con dicloroacetato in pazienti con glioblastoma di nuova diagnosi. Disponibile online: https://ClinicalTrials.gov/show/NCT00703859 (consultato il 13 luglio 2020).
<br><span id=”37″ class=”referencess blue-text”>37</span> Studio del DCA (dicloroacetato) in combinazione con cisplatino e radiazione definitiva nel carcinoma della testa e del collo. Disponibile online: https://ClinicalTrials.gov/show/NCT01386632 (consultato il 13 luglio 2020).
<br><span id=”38″ class=”referencess blue-text”>38</span> Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: Una piattaforma aperta per l’esplorazione di dati multidimensionali sulla genomica del cancro. Cancer Discov. 2012, 2, 401-404. [CrossRef] [PubMed]
<br><span id=”39″ class=”referencess blue-text”>39</span> Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6. [CrossRef] [PubMed]
<br><span id=”40″ class=”referencess blue-text”>40</span> Ragnum, H.B.; Vlatkovic, L.; Lie, A.K.; Axcrona, K.; Julin, C.H.; Frikstad, K.M.; Hole, K.H.; Seierstad, T.; Lyng, H. The tumour hypoxia marker pimonidazole reflects a transcriptional programme associated with aggressive prostate cancer. Br. J. Cancer 2015, 112, 382-390. [CrossRef]
<br><span id=”41″ class=”referencess blue-text”>41</span> Buffa, F.M.; Harris, A.L.; West, C.M.; Miller, C.J. Large meta-analysis of multiple cancers reveals a common, compact and highly prognostic hypoxia metagene. Br. J. Cancer 2010, 102, 428-435. [CrossRef]
<br><span id=”42″ class=”referencess blue-text”>42</span> Zhang, W.; Zhang, S.L.; Hu, X.; Tam, K.Y. Targeting Tumor Metabolism for Cancer Treatment: La piruvato deidrogenasi chinasi (PDK) è un valido bersaglio antitumorale? Int. J. Biol. Sci. 2015, 11, 1390-1400. [CrossRef]
<br><span id=”43″ class=”referencess blue-text”>43</span> Tang, L.; Wei, F.; Wu, Y.; He, Y.; Shi, L.; Xiong, F.; Gong, Z.; Guo, C.; Li, X.; Deng, H.; et al. Role of metabolism in cancer cell radioresistance and radiosensitization methods. J. Exp. Clin. Cancer Res. 2018, 37, 1-15. [CrossRef]
<br><span id=”44″ class=”referencess blue-text”>44</span> Stacpoole, P.W. Therapeutic Targeting of the Pyruvate Dehydrogenase Complex/Pyruvate Dehydrogenase Kinase (PDC/PDK) Axis in Cancer. J. Natl. Cancer Inst. 2017, 109. [CrossRef]
<br><span id=”45″ class=”referencess blue-text”>45</span> Saunier, E.; Benelli, C.; Bortoli, S. The Pyruvate Dehydrogenase Complex in Cancer: An Old Metabolic Gatekeeper Regulated by New Pathways and Pharmacological Agents. Int. J. Cancer 2016, 138, 809-817. [CrossRef]
<br><span id=”46″ class=”referencess blue-text”>46</span> Kolobova, E.; Tuganova, A.; Boulatnikov, I.; Popov, K.M. Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. Biochem. J. 2001, 358, 69-77. [CrossRef] [PubMed]
<br><span id=”47″ class=”referencess blue-text”>47</span> Corbet, C.; Pinto, A.; Martherus, R.; de Jesus, J.P.S.; Polet, F.; Feron, O. Acidosis Drives the Reprogramming of Fatty Acid Metabolism in Cancer Cells through Changes in Mitochondrial and Histone Acetylation. Cell Metab. 2016, 24, 311-323. [CrossRef]
<br><span id=”48″ class=”referencess blue-text”>48</span> Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding theWarburg effect: I requisiti metabolici della proliferazione cellulare. Science 2009, 324, 1029-1033. [CrossRef] [PubMed]
<br><span id=”49″ class=”referencess blue-text”>49</span> Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. Un asse mitocondriale-K+ channel è soppresso nel cancro e la sua normalizzazione promuove l’apoptosi e inibisce la crescita del cancro. Cancer Cell 2007, 11, 37-51. [CrossRef] [PubMed]
<br><span id=”50″ class=”referencess blue-text”>50</span> Woolbright, B.L.; Choudhary, D.; Mikhalyuk, A.; Trammel, C.; Shanmugam, S.; Abbott, E.; Pilbeam, C.C.; Taylor, J.A. The Role of Pyruvate Dehydrogenase Kinase-4 (PDK4) in Bladder Cancer and Chemoresistance. Mol. Cancer Ther. 2018, 17, 2004-2012. [CrossRef] [PubMed]
<br><span id=”51″ class=”referencess blue-text”>51</span> Okamoto, S.; Narita, T.; Sasanuma, H.; Takeda, S.; Masunaga, S.-I.; Bessho, T.; Tano, K. Impact of DNA repair pathways on the cytotoxicity of piperlongumine in chicken DT40 cell-lines. Genes Cancer 2014, 5, 285-292. [CrossRef] [PubMed]
<br><span id=”52″ class=”referencess blue-text”>52</span> Sun, X.; Wang, M.; Yu, X.; Guo, J.; Sun, T.; Li, X.; Yao, L.; Dong, H.; Xu, Y. Metabolic Reprogramming in Triple-Negative Breast Cancer. Front. Oncol. 2020, 10, 428. [CrossRef]
<br><span id=”53″ class=”referencess blue-text”>53</span> Lu, C.-W.; Lin, S.-C.; Chien, C.-W.; Lin, S.-C.; Lee, C.-T.; Lin, B.-W.; Lee, J.-C.; Tsai, S.-J. Overexpression of pyruvate dehydrogenase kinase 3 increases drug resistance and early recurrence in colon cancer. Am. J. Pathol. 2011, 179, 1405-1414. [CrossRef]
<br><span id=”54″ class=”referencess blue-text”>54</span> Jha, M.K.; Suk, K. Pyruvate dehydrogenase kinase as a potential therapeutic target for malignant gliomas. Brain Tumor Res. Treat. 2013, 1, 57-63. [CrossRef]
<br><span id=”55″ class=”referencess blue-text”>55</span> Blouin, J.M.; Penot, G.; Collinet, M.; Nacfer, M.; Forest, C.; Laurent-Puig, P.; Coumoul, X.; Barouki, R.; Benelli, C.; Bortoli, S. Butyrate elicits a metabolic switch in human colon cancer cells by targeting the pyruvate dehydrogenase complex. Int. J. Cancer 2011, 128, 2591-2601. [CrossRef]
<br><span id=”56″ class=”referencess blue-text”>56</span> Hur, H.; Xuan, Y.; Kim, Y.B.; Lee, G.; Shim, W.; Yun, J.; Ham, I.H.; Han, S.U. Expression of pyruvate dehydrogenase kinase-1 in gastric cancer as a potential therapeutic target. Int. J. Oncol. 2013, 42, 44-54. [CrossRef] [PubMed]
<br><span id=”57″ class=”referencess blue-text”>57</span> Wigfield, S.M.; Winter, S.C.; Giatromanolaki, A.; Taylor, J.; Koukourakis, M.L.; Harris, A.L. PDK-1 regola la produzione di lattato in ipossia ed è associata a una prognosi sfavorevole nel cancro squamoso della testa e del collo. Br. J. Cancer 2008, 98, 1975-1984. [CrossRef] [PubMed]
<br><span id=”58″ class=”referencess blue-text”>58</span> Sun, W.; Zhou, S.; Chang, S.S.; McFate, T.; Verma, A.; Califano, J.A. Mutazioni mitocondriali contribuiscono all’accumulo di HIF1alfa attraverso l’aumento delle specie reattive dell’ossigeno e l’up-regolazione della piruvato deidrogenasi chinasi 2 nel carcinoma a cellule squamose della testa e del collo. Clin. Cancer Res. 2009, 15, 476-484. [CrossRef] [PubMed]
<br><span id=”59″ class=”referencess blue-text”>59</span> Shen, Y.C.; Ou, D.L.; Hsu, C.; Lin, K.L.; Chang, C.Y.; Lin, C.Y.; Liu, S.H.; Cheng, A.L. Activating oxidative phosphorylation by a pyruvate dehydrogenase kinase inhibitor overcomes sorafenib resistance of hepatocellular carcinoma. Br. J. Cancer 2013, 108, 72-81. [CrossRef] [PubMed]
<br><span id=”60″ class=”referencess blue-text”>60</span> Fujiwara, S.; Kawano, Y.; Yuki, H.; Okuno, Y.; Nosaka, K.; Mitsuya, H.; Hata, H. PDK1 inhibition is a novel therapeutic target in multiple myeloma. Br. J. Cancer 2013, 108, 170-178. [CrossRef]
<br><span id=”61″ class=”referencess blue-text”>61</span> Guda, M.R.; Asuthkar, S.; Labak, C.M.; Tsung, A.J.; Alexandrov, I.; Mackenzie, M.J.; Prasad, D.V.; Velpula, K.K. Targeting PDK4 inhibits breast cancer metabolism. Am. J. Cancer Res. 2018, 8, 1725-1738.
<br><span id=”62″ class=”referencess blue-text”>62</span> Roh, J.L.; Park, J.Y.; Kim, E.H.; Jang, H.J.; Kwon, M. L’attivazione dell’ossidazione mitocondriale mediante l’inibizione di PDK2 inverte la resistenza al cisplatino nel cancro della testa e del collo. Cancer Lett. 2016, 371, 20-29. [CrossRef]
<br><span id=”63″ class=”referencess blue-text”>63</span> Dupuy, F.; Tabariès, S.; Andrzejewski, S.; Dong, Z.; Blagih, J.; Annis, M.G.; Omeroglu, A.; Gao, D.; Leung, S.; Amir, E.; et al. PDK1-Dependent Metabolic Reprogramming Dictates Metastatic Potential in Breast Cancer. Cell Metab. 2015, 22, 577-589. [CrossRef]
<br><span id=”64″ class=”referencess blue-text”>64</span> McFate, T.; Mohyeldin, A.; Lu, H.; Thakar, J.; Henriques, J.; Halim, N.D.; Wu, H.; Schell, M.J.; Tsang, T.M.; Teahan, O.; et al. L’attività del complesso della piruvato deidrogenasi controlla il fenotipo metabolico e maligno nelle cellule tumorali. J. Biol. Chem. 2008, 283, 22700-22708. [CrossRef]
<br><span id=”65″ class=”referencess blue-text”>65</span> Shavit, R.; Ilouze, M.; Feinberg, T.; Lawrence, Y.R.; Tzur, Y.; Peled, N. Mitochondrial induction as a potential radio-sensitizer in lung cancer cells-A short report. Cell. Oncol. 2015, 38, 247-252. [CrossRef]
<br><span id=”66″ class=”referencess blue-text”>66</span> Allen, K.T.; Chin-Sinex, H.; DeLuca, T.; Pomerening, J.; Sherer, J.; Watkins, J.; Foley, J.; Jesseph, J.; Mendonca, M. Il dicloroacetato altera il metabolismo di Warburg, inibisce la crescita cellulare e aumenta la sensibilità ai raggi X delle cellule tumorali polmonari umane A549 e H1299 NSC. Free Radic. Biol. Med. 2015, 89, 263-273. [CrossRef] [PubMed]
<br><span id=”67″ class=”referencess blue-text”>67</span> Sun, L.; Moritake, T.; Ito, K.; Matsumoto, Y.; Yasui, H.; Nakagawa, H.; Hirayama, A.; Inanami, O.; Tsuboi, K. Metabolic analysis of radioresistant medulloblastoma stem-like clones and potential therapeutic targets. PLoS ONE 2017, 12, e0176162. [CrossRef] [PubMed]
<br><span id=”68″ class=”referencess blue-text”>68</span> Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Iniziatori, amplificatori o tallone d’Achille? Nat. Rev. Cancer 2014, 14, 709-721. [CrossRef] [PubMed]
<br><span id=”69″ class=”referencess blue-text”>69</span> Feng, H.; Wang, J.; Chen, W.; Shan, B.; Guo, Y.; Xu, J.; Wang, L.; Guo, P.; Zhang, Y. Hypoxia-induced autophagy as an additional mechanism in human osteosarcoma radioresistance. J. Bone Oncol. 2016, 5, 67-73. [CrossRef]
<br><span id=”70″ class=”referencess blue-text”>70</span> Samanta, D.; Park, Y.; Andrabi, S.A.; Shelton, L.M.; Gilkes, D.M.; Semenza, G.L. PHGDH Expression Is Required for Mitochondrial Redox Homeostasis, Breast Cancer Stem Cell Maintenance, and Lung Metastasis. Cancer Res. 2016, 76, 4430-4442. [CrossRef]
<br><span id=”71″ class=”referencess blue-text”>71</span> Diepart, C.; Karroum, O.; Magat, J.; Feron, O.; Verrax, J.; Calderon, P.B.; Grégoire, V.; Leveque, P.; Stockis, J.; Dauguet, N.; et al. Il trattamento con triossido di arsenico diminuisce il tasso di consumo di ossigeno delle cellule tumorali e radiosensibilizza i tumori solidi. Cancer Res. 2012, 72, 482-490. [CrossRef]
<br><span id=”72″ class=”referencess blue-text”>72</span> Maggiorella, L.; Wen, B.; Frascogna, V.; Opolon, P.; Bourhis, J.; Deutsch, E. Effetti combinati di sensibilizzazione alle radiazioni e antiangiogenici delle radiazioni ionizzanti e dell’inibitore della proteasi ritonavir in un modello di carcinoma della testa e del collo. Anticancer Res. 2005, 25, 4357-4362.
<br><span id=”73″ class=”referencess blue-text”>73</span> Dwarakanath, B.S. Citotossicità, radiosensibilizzazione e chemiosensibilizzazione delle cellule tumorali da parte del 2-deossi-D-glucosio in vitro. J. Cancer Res. Ther. 2009, 5 (Suppl. 1), 27-31. [CrossRef]
<br><span id=”74″ class=”referencess blue-text”>74</span> Kim, J.H.; Kim, S.H.; He, S.Q.; Alfieri, A.A.; Young, C.W. Potenziamento degli effetti delle radiazioni su sferoidi tumorali multicellulari (MTS) di cellule HeLa da parte della lonidamina. Int. J. Radiat. Oncol. Biol. Phys. 1989, 16, 1277-1280. [CrossRef]
<br><span id=”75″ class=”referencess blue-text”>75</span> Nath, K.; Guo, L.; Nancolas, B.; Nelson, D.S.; Shestov, A.A.; Lee, S.C.; Roman, J.; Zhou, R.; Leeper, D.B.; Halestrap, A.P.; et al. Mechanism of antineoplastic activity of lonidamine. Biochim. Biophys Acta 2016, 1866, 151-162. [CrossRef]
<br><span id=”76″ class=”referencess blue-text”>76</span> Blackburn, A.C.; Matthaei, K.I.; Lim, C.; Taylor, M.C.; Cappello, J.Y.; Hayes, J.D.; Anders, M.W.; Board, P.G. Deficiency of glutathione transferase zeta causes oxidative stress and activation of antioxidant response pathways. Mol. Pharmacol. 2006, 69, 650-657. [CrossRef] [PubMed]
<br><span id=”77″ class=”referencess blue-text”>77</span> Theodoratos, A.; Tu, W.J.; Cappello, J.; Blackburn, A.C.; Matthaei, K.; Board, P.G. Phenylalanine-induced leucopenia in genetic and dichloroacetic acid generated deficiency of glutathione transferase Zeta. Biochem. Pharmacol. 2009, 77, 1358-1363. [CrossRef] [PubMed]
<br><span id=”78″ class=”referencess blue-text”>78</span> Michelakis, E.D.; Webster, L.; Mackey, J.R. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br. J. Cancer 2008, 99, 989-994. [CrossRef] [PubMed]
<br><span id=”79″ class=”referencess blue-text”>79</span> Stacpoole, P.W. The pharmacology of dichloroacetate. Metabolismo 1989, 38, 1124-1144. [CrossRef]
<br><span id=”80″ class=”referencess blue-text”>80</span> Halestrap, A.P. The mitochondrial pyruvate carrier. Cinetica e specificità per substrati e inibitori. Biochem. J. 1975, 148, 85-96. [CrossRef]
<br><span id=”81″ class=”referencess blue-text”>81</span> Pathak, R.K.; Marrache, S.; Harn, D.A.; Dhar, S. Mito-DCA: un’impalcatura molecolare mirata ai mitocondri per la somministrazione efficace del modulatore metabolico dicloroacetato. ACS Chem. Biol. 2014, 9, 1178-1187. [CrossRef]
<br><span id=”82″ class=”referencess blue-text”>82</span> Gang, B.P.; Dilda, P.J.; Hogg, P.J.; Blackburn, A.C. Targeting of two aspects of metabolism in breast cancer treatment. Cancer Biol. Ther. 2014, 15, 1533-1541. [CrossRef]
<br><span id=”83″ class=”referencess blue-text”>83</span> Sutendra, G.; Dromparis, P.; Kinnaird, A.; Stenson, T.H.; Haromy, A.; Parker, J.M.R.; McMurtry, M.S.; Michelakis, E.D. Mitochondrial activation by inhibition of PDKII suppresses HIF1a signaling and angiogenesis in cancer. Oncogene 2013, 32, 1638-1650. [CrossRef]
<br><span id=”84″ class=”referencess blue-text”>84</span> Xuan, Y.; Hur, H.; Ham, I.H.; Yun, J.; Lee, J.Y.; Shim, W.; Kim, Y.B.; Lee, G.; Han, S.U.; Cho, Y.K. Il dicloroacetato attenua la resistenza al 5-fluorouracile indotta dall’ipossia nel cancro gastrico attraverso la regolazione del metabolismo del glucosio. Exp. Cell Res. 2014, 321, 219-230. [CrossRef]
<br><span id=”85″ class=”referencess blue-text”>85</span> Cesi, G.; Walbrecq, G.; Zimmer, A.; Kreis, S.; Haan, C. La produzione di ROS indotta dal trattamento con l’inibitore di BRAF ricrea i processi metabolici che influenzano la crescita cellulare delle cellule di melanoma. Mol. Cancer 2017, 16, 1-16. [CrossRef]
<br><span id=”86″ class=”referencess blue-text”>86</span> Kluza, J.; Corazao-Rozas, P.; Touil, Y.; Jendoubi, M.; Maire, C.; Guerreschi, P.; Jonneaux, A.; Ballot, C.; Balayssac, S.l’inattivazione dell’asse di segnalazione HIF-1α/PDK3 spinge il melanoma verso il metabolismo ossidativo mitocondriale e potenzia l’attività terapeutica dei pro-ossidanti. Cancer Res. 2012, 72, 5035-5047. [CrossRef] [PubMed]
<br><span id=”87″ class=”referencess blue-text”>87</span> Sun, H.; Zhu, A.; Zhou, X.; Wang, F. La soppressione della piruvato deidrogenasi chinasi-2 risensibilizza le cellule di cancro al polmone umano resistenti al paclitaxel. Oncotarget 2017, 8, 52642-52650. [CrossRef] [PubMed]
<br><span id=”88″ class=”referencess blue-text”>88</span> Velpula, K.K.; Bhasin, A.; Asuthkar, S.; Tsung, A.J. Combined targeting of PDK1 and EGFR triggers regression of glioblastoma by reversing the Warburg effect. Cancer Res. 2013, 73, 7277-7289. [CrossRef] [PubMed]
<br><span id=”89″ class=”referencess blue-text”>89</span> Zhang, W.; Hu, X.; Zhou, W.; Tam, K.Y. Liquid Chromatography-Tandem Mass Spectrometry Method Revealed that Lung Cancer Cells Exhibited Distinct Metabolite Profiles upon the Treatment with Different Pyruvate Dehydrogenase Kinase Inhibitors. J. Proteome Res. 2018, 17, 3012-3021. [CrossRef]
<br><span id=”90″ class=”referencess blue-text”>90</span> Dubuis, S.; Ortmayr, K.; Zampieri, M. A framework for large-scale metabolome drug profiling links coenzyme A metabolism to the toxicity of anti-cancer drug dichloroacetate. Commun. Biol. 2018, 1, 1-11. [CrossRef]
<br><span id=”91″ class=”referencess blue-text”>91</span> El Sayed, S.M.; Baghdadi, H.; Ahmed, N.S.; Almaramhy, H.H.; Mahmoud, A.A.; El-Sawy, S.A.; Ayat, M.elshazley, M.; Abdel-Aziz, W.; Abdel-Latif, H.M.; et al. Il dicloroacetato è un antimetabolita che antagonizza l’acetato e priva le cellule tumorali dei suoi benefici: Una nuova ipotesi medica basata sull’evidenza. Med. Hypotheses 2019, 122, 206-209. [CrossRef]
<br><span id=”92″ class=”referencess blue-text”>92</span> Li, X.; Liu, J.; Hu, H.; Lu, S.; Lu, Q.; Quan, N.; Rousselle, T.; Patel, M.S.; Li, J. Dichloroacetate Ameliorates Cardiac Dysfunction Caused by Ischemic Insults Through AMPK Signal Pathway-Not Only Shifts Metabolism. Toxicol. Sci. 2019, 167, 604-617. [CrossRef]
<br><span id=”93″ class=”referencess blue-text”>93</span> Li, W.; Saud, S.M.; Young, M.R.; Chen, G.; Hua, B. Targeting AMPK for cancer prevention and treatment. Oncotarget 2015, 6, 7365-7378. [CrossRef]
<br><span id=”94″ class=”referencess blue-text”>94</span> Dunbar, E.M.; Coats, B.S.; Shroads, A.L.; Langaee, T.; Lew, A.; Forder, J.R.; Shuster, J.J.; Wagner, D.A.; Stacpoole, P.W. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Investig. New Drugs 2014, 32, 452-464. [CrossRef]
<br><span id=”95″ class=”referencess blue-text”>95</span> Michelakis, E.D.; Sutendra, G.; Dromparis, P.; Webster, L.; Haromy, A.; Niven, E.; Maguire, C.; Gammer, T.L.; Mackey, J.R.; Fulton, D.; et al. Modulazione metabolica del glioblastoma con dicloroacetato. Sci. Transl. Med. 2010, 2. [CrossRef]
<br><span id=”96″ class=”referencess blue-text”>96</span> De Ridder, M.; Verovski, V.N.; Chiavaroli, C.; Berge, D.L.V.D.; Monsaert, C.; Law, K.; Storme, G.A. L’effetto radiosensibilizzante dell’immunoadiuvante OM-174 richiede la cooperazione tra cellule immunitarie e tumorali attraverso l’interferone-gamma e l’ossido nitrico sintasi inducibile. Int. J. Radiat. Oncol. Biol. Phys. 2006, 66, 1473-1480. [CrossRef] [PubMed]
<br><span id=”97″ class=”referencess blue-text”>97</span> Wang, H.; Bouzakoura, S.; de Mey, S.; Jiang, H.; Law, K.; Dufait, I.; Corbet, C.; Verovski, V.; Gevaert, T.; Feron, O.; et al. Auranofin radiosensibilizza le cellule tumorali attraverso il bersaglio della tioredoxina reduttasi e la conseguente sovrapproduzione di specie reattive dell’ossigeno. Oncotarget 2017, 8, 35728-35742. [CrossRef] [PubMed]
<br><span id=”98″ class=”referencess blue-text”>98</span> de Mey, S.; Jiang, H.; Corbet, C.; Wang, H.; Dufait, I.; Law, K.; Bastien, E.; Verovski, V.; Gevaert, T.; Feron, O.; et al. Antidiabetic Biguanides Radiosensitize Hypoxic Colorectal Cancer Cells Through a Decrease in Oxygen Consumption. Front. Pharmacol. 2018, 9, 1073. [CrossRef] [PubMed]
<br><span id=”99″ class=”referencess blue-text”>99</span> Noeparast, A.; Teugels, E.; Giron, P.; Verschelden, G.; De Brakeleer, S.; Decoster, L.; De Grève, J. Le mutazioni BRAF non-V600 ricorrenti nel tumore del polmone predicono la sensibilità alla combinazione di Trametinib e Dabrafenib. Oncotarget 2016, 8, 60094-60108. [CrossRef]
<br><span id=”100″ class=”referencess blue-text”>100</span> Polet, F.; Corbet, C.; Pinto, A.; Rubio, L.I.; Martherus, R.; Bol, V.; Drozak, X.; Grégoire, V.; Riant, O.; Feron, O. La riduzione della disponibilità di serina integra l’inibizione del metabolismo della glutammina per bloccare la crescita delle cellule leucemiche. Oncotarget 2015, 7, 1765-1776. [CrossRef]
<p></p>
<p>Contenuto correlato:</p>
<figure class=”wp-block-embed is-type-wp-embed is-provider-dca-guide wp-block-embed-dca-guide”><div class=”wp-block-embed__wrapper”>
</div></figura>
<figure class=”wp-block-embed is-type-wp-embed is-provider-dca-guide wp-block-embed-dca-guide”><div class=”wp-block-embed__wrapper”>
</div></figura>