Jason Y.Y. Wong1, Gordon S. Huggins2, Marcella Debidda4, Nikhil C. Munshi4 e Immacolata De Vivo1,3
1 Laboratorio Channing, Dipartimento di Medicina, Brigham and Women’s Hospital e Harvard Medical School, Boston Massachusetts.
2 Istituto di ricerca sulla cardiologia molecolare, Tufts-New England Medical Center, Boston Massachusetts.
3 Programma di epidemiologia molecolare e genetica, Harvard School of Public Health, Boston Massachusetts.
4 Jerome Lipper Multiple Myeloma Center, Department of Medical Oncology, Dana-Farber Cancer Institute, Harvard Medical School, Boston Massachusetts.
Corrispondenza: Immacolata De Vivo, Brigham and Women’s Hospital, Dipartimento di Medicina, Channing Laboratory, 181 Longwood Ave, Boston, MA, 02115, USA. Telefono: 617-525-2094. Fax: 617-525-2008. [email protected].
Ricevuto: 14 settembre 2020
Accettato: 4 dicembre 2020
Pubblicato: 9 dicembre 2020
Abstract
Scopo: Un recente studio di riferimento ha dimostrato che il trattamento con dicloroacetato (DCA) promuove l’apoptosi in linee cellulari di cancro del polmone, della mammella e del glioblastoma, spostando il metabolismo dalla glicolisi aerobica all’ossidazione del glucosio, insieme al rimodellamento dell’asse NFAT-Kv1.5. L’obiettivo di questo studio è stato quello di determinare se il DCA induce l’apoptosi nelle cellule di cancro dell’endometrio e di valutare il meccanismo apoptotico.
Metodi: Un gruppo di linee cellulari di cancro endometriale con vari gradi di differenziazione è stato trattato con DCA e analizzato per l’apoptosi mediante citometria a flusso. Per valutare il meccanismo apoptotico sono stati esaminati i correlati biologici come l’espressione genica, il Ca2+ intracellulare e il potenziale di membrana mitocondriale.
Risultati: L’avvio dell’apoptosi è stato osservato in cinque linee cellulari tumorali a bassa o moderata invasività, tra cui Ishikawa, RL95-2, KLE, AN3CA e SKUT1B, mentre il trattamento non ha avuto alcun effetto sulle cellule 293T non cancerose. Due linee cellulari di adenocarcinoma endometriale altamente invasive, HEC1A e HEC1B, sono risultate resistenti all’apoptosi indotta dal DCA. Le linee cellulari che rispondevano all’apoptosi presentavano un aumento significativo dell’apoptosi precoce e tardiva, una diminuzione del potenziale di membrana mitocondriale e una riduzione dell’abbondanza del trascritto di Survivin, coerenti con un meccanismo regolato dai mitocondri. Il trattamento con DCA ha diminuito i livelli di calcio intracellulare nella maggior parte delle linee cellulari che rispondono all’apoptosi, il che suggerisce un contributo della via mediata da NFAT-Kv1.5. Il trattamento con DCA ha aumentato i trascritti del modulatore dell’apoptosi p53 upregolato (PUMA) nelle linee cellulari con risposta apoptotica, suggerendo il coinvolgimento di un meccanismo mediato da p53-PUMA.
Parole chiave: Dicloroacetato; Endometrio; Cancro; Apoptosi; Mitocondri
Dichiarazione di conflitto di interessi: Gli autori dichiarano di non avere conflitti di interesse.
Conclusioni: Il dicloroacetato sensibilizza efficacemente la maggior parte delle linee cellulari di cancro endometriale all’apoptosi attraverso meccanismi mitocondriali, NFAT-Kv1.5 e PUMA-mediati. Sono giustificate ulteriori indagini sul potenziale terapeutico antitumorale del DCA.
La MMP è ulteriormente confermata dalla colorazione fluorescente TMRM in un esperimento di risposta alla dose di DCA. Le barre di errore rappresentano l’errore standard di 2 esperimenti indipendenti eseguiti in pozzetti triplicati.
INTRODUZIONE
Il cancro dell’endometrio (EC) è una neoplasia del rivestimento epiteliale del corpo uterino. È la neoplasia ginecologica più comune negli Stati Uniti e la quarta causa di morte per cancro nel Paese tra le donne [1]. Esistono poche opzioni terapeutiche prive di gravi inconvenienti per le persone affette da tumore dell’endometrio ricorrente o metastatico. La chemioterapia per la malattia metastatica presenta alti tassi di tossicità, nevralgie e complicazioni cardiache[2, 3]. L’impulso allo sviluppo di future terapie antitumorali sarà quello di ridurre i gravi effetti avversi, dimostrando al contempo un’efficacia paragonabile o migliore rispetto ai trattamenti esistenti.
La glicolisi aerobica, nota anche come “effetto Warburg”, è una proprietà unica della maggior parte dei tumori. Questo fenomeno è caratterizzato da un aumento dell’assorbimento di glucosio e dalla dipendenza dalla glicolisi per la produzione di ATP nonostante una fonte di ossigeno disponibile [5]. Si ritiene che la glicolisi aerobica sia il risultato di una disfunzione mitocondriale che conferisce resistenza apoptotica alle cellule tumorali [6]. Questa resistenza apoptotica è dovuta all’iperpolarizzazione della membrana mitocondriale che impedisce il rilascio di mediatori pro-apoptotici dai mitocondri al citoplasma [4]. Le membrane mitocondriali iperpolarizzate sono caratteristiche della maggior parte dei carcinomi e la loro inversione è associata all’avvio dell’apoptosi [7,8].
Il bersaglio terapeutico della glicolisi aerobica è un mezzo nuovo per colpire le cellule tumorali. Il regolatore chiave del metabolismo cellulare è la piruvato deidrogenasi (PDH), che a sua volta è inibita dalla piruvato deidrogenasi chinasi (PDK). Uno studio recente ha dimostrato che l’attività della PDK nelle linee cellulari tumorali può essere ridotta dal DCA [4]. Il bersaglio metabolico del DCA coinvolge due meccanismi sinergici, la via prossimale e quella distale [4]. Nella via prossimale (regolata dai mitocondri), il DCA si lega alla PDK e attenua l’inibizione dell’attività della PDH. L’aumento dell’attività della PDH sposta il metabolismo dalla glicolisi all’ossidazione del glucosio e diminuisce l’iperpolarizzazione del potenziale di membrana mitocondriale (MMP), che apre i pori di transizione mitocondriali (MTP). Ciò consente la traslocazione delle specie reattive dell’ossigeno (ROS) e del citocromo c dai mitocondri al citoplasma, inducendo successivamente l’apoptosi attraverso l’attivazione delle caspasi [4]. Nella via distale (NFAT-Kv1.5), i ROS traslocati dilatano i canali degli ioni potassio Kv1.5 sulla membrana plasmatica. L’espulsione di ioni potassio iperpolarizza la cellula, impedendo l’ingresso di Ca2+ voltaggio-dipendente. La diminuzione del livello di Ca2+ intracellulare inibisce l’attivazione di NFAT, che aumenta ulteriormente l’espressione di Kv1.5, creando un circuito di feedback positivo che si traduce in una riduzione dell’inibizione tonica delle caspasi [4].
Esiste un sostanziale cross-talk tra le vie apoptotiche mitocondriali e quelle mediate da p53. La p53-upregulatedmodulator of apoptosis (PUMA) è una di queste proteine pro-apoptotiche che fa da ponte tra i meccanismi mitocondriali e quelli mediati dal soppressore tumorale p53. PUMA è un membro della famiglia di proteine BH3-only la cui espressione è regolata trascrizionalmente da p53 [9-11]. In seguito all’attivazione da parte di vari stimoli apoptotici, PUMA trasloca sulla membrana mitocondriale dove antagonizza le proteine Bcl-2 pro-sopravvivenza legandosi al suo dominio BH3, inducendo il rilascio di citocromo c e promuovendo l’apoptosi [10,12]. In recenti studi di knock-out, è stato determinato che PUMA è un mediatore critico dell’apoptosi p53-dipendente nei timociti murini e nelle cellule umane di cancro colorettale [13,14].
In numerosi studi è stato dimostrato che il dicloroacetato promuove l’ossidazione del glucosio in vari disturbi mitocondriali [15,16]. Inoltre, negli studi clinici sulle encefalomiopatie mitocondriali il trattamento con DCA è risultato avere effetti collaterali più lievi rispetto a quelli delle attuali terapie per il cancro dell’endometrio [17]. Ad oggi, l’effetto del DCA è stato studiato in un numero limitato di linee cellulari tumorali e la nostra comprensione dei meccanismi apoptotici alternativi regolati dal DCA è carente. Lo scopo del nostro studio è stato quello di determinare se il DCA sensibilizza un gruppo di linee cellulari di cancro endometriale all’apoptosi e di valutare il contributo dei meccanismi regolati dai mitocondri, NFAT-Kv1.5 e PUMA nel processo apoptotico esaminando i correlati biologici.
Materiali e metodi
Coltura cellulare
Le linee cellulari AN3CA, SKUT1B, RL95-2, KLE, HEC1A e HEC1B sono state acquistate dall’American Type Culture Collection (Manassas, VA) e la linea cellulare Ishikawa è stata acquistata da Sigma-Aldrich (St. Louis, MO). Le cellule epiteliali renali 293T, che servivano come controlli sani non cancerosi, sono state fornite da Nikhil Munshi. L’adenocarcinoma epiteliale mammario MCF7 è stato un dono di Ramon Parsons (Columbia University). Le linee cellulari sono state propagate secondo le condizioni specificate dal distributore. Le linee cellulari sono state mantenute in un incubatore umidificato a 37°C, 5%CO2. I terreni di crescita DMEM, McCoy’s 5A, MEM e DMEM-F12, insieme ai supplementi di penicillina-streptomicina e insulina, sono stati acquistati da Gibco-Invitrogen (Carlsbad, CA). Il dicloroacetato (Alfa Aesar, Ward Hill, MA) è stato disciolto in una soluzione di lavoro 1M, sterilizzato in filtro e successivamente diluito alle concentrazioni di trattamento nei terreni di crescita.
Test di vitalità cellulare
La vitalità cellulare è stata misurata utilizzando il reagente CellTiter-Blue (Promega) che misura la capacità delle cellule sane vitali di metabolizzare un substrato di Resazurin in un prodotto fluorescente di Resorufin. In breve, 3×104 cellule di ciascuna linea cellulare sono state piastrate in piastre di coltura a 96 pozzetti a pareti opache e incubate in condizioni di crescita standard per una notte fino al 60-70% di confluenza. Il terreno di coltura in ogni pozzetto è stato quindi sostituito con terreno di coltura fresco contenente concentrazioni crescenti di DCA (0 mM, 1 mM, 5 mM, 10 mM). Ogni pozzetto è stato eseguito in triplicato in due o più esperimenti indipendenti per ciascuna linea cellulare. Dopo il trattamento, le piastre sono state incubate per 40 ore a 37°C, dopodiché sono stati aggiunti 20 µL di substrato di Resazurin direttamente in ogni pozzetto e incubati per altre 3 ore. Le piastre sono state quindi lette su un lettore di piastre Molecular Devices Gemini XPS (Sunnyvale, CA) a 560/590 nm.
Saggi di apoptosi
Per determinare se il trattamento induce specificamente un’apoptosi precoce è stata utilizzata la citometria a flusso con colorazione Annexin-V-FITC (BD Bioscience, San Jose, CA) e 7-amino-actinomicina D (7-AAD). In breve, 5×105 cellule per ciascuna linea cellulare sono state seminate in piastre di coltura tissutale a 6 pozzetti e incubate per una notte fino al 60-70% di confluenza in condizioni di crescita standard. Il terreno di coltura per ciascuna linea cellulare è stato quindi sostituito con terreno di coltura fresco con e senza una dose di DCA di 10 mM. I gruppi di trattamento per ciascuna linea cellulare sono stati replicati tre volte. Le cellule sono state quindi incubate per 40 ore a 37°C e raccolte con Trypsin-EDTA 0,25% (Invitrogen, Carlsbad, CA). Le cellule sono state lavate con PBS 1X e successivamente colorate secondo il protocollo del produttore. La citometria a flusso è stata eseguita su un BD FACSCanto II (BD Bioscience) e i dati sono stati analizzati con FlowJo 7.2.2 (Tree Star, Ashland, OR) e il software BD FACSDiva 6.0 (BD Bioscience).
Il kit Apoptag Peroxidase Terminal dUTP Nick-end Labeling (TUNEL) (Millipore, Billerica, MA) è stato utilizzato per visualizzare le cellule apoptotiche che avevano subito una frammentazione genomica caspasi-dipendente. Brevemente, 5×104 cellule di diverse linee cellulari rappresentative di cancro endometriale sono state seminate e propagate su vetrini da camera a 4 pozzetti (Nunc, Rochester, NY) per una notte. Il terreno di coltura in ogni pozzetto è stato poi sostituito con terreno di coltura fresco con o senza 10 mM DCA. Dopo 48 ore, le cellule sono state fissate con paraformaldeide all’1% e colorate secondo il protocollo del produttore. La colorazione è stata eseguita dal Dana Farber – Harvard Cancer Center Pathology Core facility. Le immagini sono state acquisite con obiettivo 40X su un microscopio Zeiss Axioskop 2 Plus (Thornwood, NY) utilizzando il software AxioVs40 v.4.4.1.0 a 24 bit RGB.
Saggio di proliferazione cellulare
Per misurare la proliferazione cellulare è stata utilizzata la citometria a flusso con bromodeossiuridina (BrdU) (BD Bioscience) e colorazione 7-AAD. In breve, diverse linee cellulari rappresentative di carcinoma endometriale sono state propagate come indicato sopra per il test dell’Annexin-V. Le cellule sono state poi messe in astinenza da siero per 8 ore in terreni di crescita contenenti lo 0,5% di FBS per resettare il ciclo cellulare alla fase G0. Il terreno di coltura è stato successivamente modificato in un normale terreno di coltura con e senza trattamento con DCA 10 mM. Dopo 24 ore, le cellule sono state pulsate per 2 ore con 10 µM BrdU in terreno di crescita. Le cellule sono state quindi raccolte, colorate e analizzate secondo il protocollo del produttore.
Saggi del potenziale di membrana mitocondriale
Il potenziale di membranamitocondrialeè stato rilevato utilizzando il Mitocapture Apoptosis Detection Kit (Calbiochem). La crescita, il trattamento e il layout sperimentale delle linee cellulari sono stati identici a quelli del test di Annexin-V descritto sopra. Dopo un periodo di incubazione di 24 ore in seguito al trattamento con e senza DCA 10 mM, le cellule sono state raccolte e lavate con PBS 1X, colorate con il reagente Mitocapture secondo il protocollo del produttore e analizzate mediante citometria a flusso. Per valutare la MMP è stata utilizzata anche una versione modificata di un protocollo che prevede la colorazione dei mitocondri con tetrametil rodamina metil estere (TMRM) (Invitrogen, Carlsbad, CA), descritto altrove [18]. In breve, le cellule sono state propagate e trattate esattamente come nel saggio di vitalità cellulare descritto sopra. Dopo un periodo di incubazione di 24 ore, 5×104 cellule sono state isolate, lavate in PBS 1X e risospese in soluzione salina tamponata di Hanks (HBSS) (Sigma-Aldrich, St. Louis, MA) con 50 nM di TMRM e incubate per 30 minuti a 37 °C. Le cellule sono state trasferite in una piastra opaca a 96 pozzetti e la fluorescenza è stata misurata a 530/620 nm a 37°C utilizzando un lettore di piastre.
Livelli di calcio intracellulare
I livelli di calcio intracellulare sono stati misurati utilizzando il FLUO-4 NW Calcium Assay (Invitrogen). In breve, 3×104 cellule per ogni linea cellulare sono state placcate su piastre di coltura a 96 pozzetti a pareti opache e incubate in condizioni di crescita standard per 8 ore. Il terreno di coltura in ogni pozzetto è stato poi sostituito con terreno di coltura fresco contenente concentrazioni crescenti di DCA. Ogni gruppo di trattamento è stato replicato in 4 pozzetti in almeno 2 esperimenti indipendenti. Dopo 8 ore di incubazione, le cellule in ogni pozzetto sono state trattate con il reagente FLUO-4 secondo il protocollo del produttore. Le piastre sono state quindi lette su un lettore di piastre fluorescenti a 494/516 nm.
PCR in tempo reale
La PCR quantitativa in tempo reale è stata utilizzata per rilevare l’abbondanza dei trascritti endogeni di Survivin e PUMA. Un totale di 1×106 cellule per ogni linea cellulare sono state seminate e fatte crescere in piastre di coltura tissutale da 10 cm per una notte. Il terreno di coltura è stato poi sostituito con terreno di coltura fresco con o senza trattamento con 10 mM DCA. Dopo 40 ore di incubazione, sono state raccolte 3×106 cellule per ogni piastra e l’RNA totale è stato estratto con RNeasy Plus Mini Kit (Qiagen, Valencia, CA) secondo il protocollo del produttore. Il cDNA a primo filamento è stato sintetizzato con 1000 ng di RNA totale e primer Oligo dT utilizzando Superscript III Reverse Transcriptase (Invitrogen) come da protocollo del produttore. Il prodotto cDNA è stato poi trattato con RNasi H per 20 minuti a 37°C e diluito a 100 ng/µL. Le concentrazioni di RNA e cDNA sono state determinate con precisione utilizzando uno spettrofotometro Nanodrop ND-1000 (Wilmington, DE).
Le sequenze dei primer per la Survivin sono state forward 5′-AAGAACTGGCCCTTCTTGGA-3′ e reverse 5′- CAACCGGACGAATGCTT-3′ (Primerbank). Le sequenze dei primer per PUMA e il gene housekeeping RPLP0 sono state descritte in studi precedenti [19, 20]. Le miscele di reazione erano costituite da 1X Applied Biosystems SYBR Green PCR mix (Foster City, CA), 1,5 mM MgCl2, 0,42 mM dNTPs, 5U ABI Amplitaq Gold, 200 ng di cDNA template e 333 nM di primer forward e reverse. Le reazioni sono state eseguite in triplo in due esperimenti replicati. Le condizioni di ciclaggio erano 1 ciclo a 95C per 10:00, 33x cicli di 95C per 0:30, 55C per 0:30 e 72C a 0:30. Una curva standard a cinque punti per le reazioni ha avuto pendenze lineari di -3,2 +/- 0,1 con coefficienti di correlazione (r2) superiori a 0,985. Il test è stato eseguito con un sistema ABI 7300 Real-Time PCR (Foster City, CA). La quantificazione relativa dei trascritti target normalizzati a RPLP0 è stata valutata con il software di studio RQ di ABI 7300 Real-Time PCR Systems utilizzando il metodo Ct comparativo.
Analisi statistica
Per valutare le differenze tra i bracci di trattamento sono stati utilizzati il test t di Student e l’ANOVA a una via. Un valore di p inferiore a 0,05 è stato considerato significativo. L’analisi è stata eseguita con Microsoft Excel 2007 (Redmond, WA). I grafici sono stati creati con GraphPad Prism 5 (San Diego, CA).
Risultati
IlDCA riduce la vitalità delle cellule di cancro endometriale in modo dose-dipendente
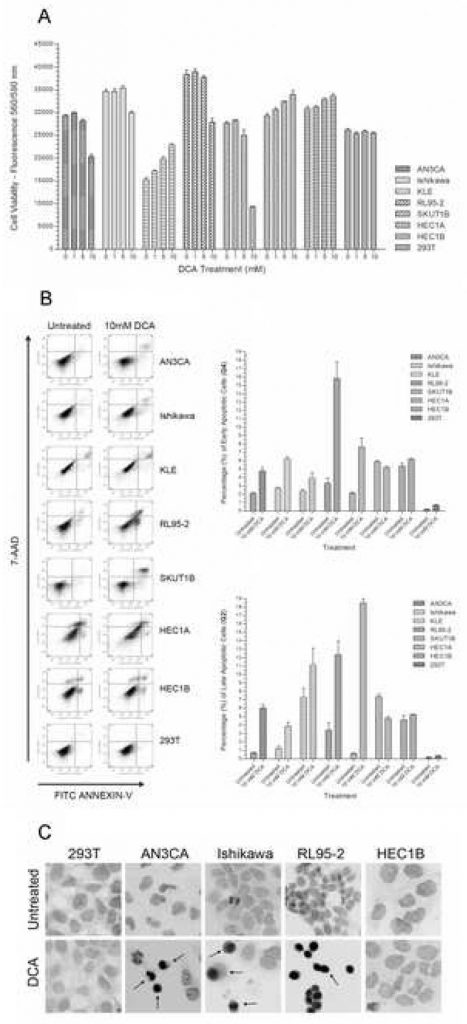
Per determinare l’effetto del DCA sulla vitalità delle cellule di cancro endometriale, ogni linea cellulare è stata coltivata con dosi crescenti di DCA. In un gruppo di sette linee cellulari di cancro endometriale, AN3CA, Ishikawa, RL95-2 e SKUT1B hanno registrato una riduzione della vitalità compresa tra il 15% e il 75% con l’aumento della concentrazione di DCA (Figura 1A). La riduzione della vitalità per la maggior parte delle linee cellulari rispondenti ha raggiunto la significatività alla dose di 10 mM. Il confronto tra il gruppo non trattato e il gruppo trattato con DCA 10 mM ha avuto valori di p < 0,01 per AN3CA, Ishikawa, RL95-2 e SKUT1B. Una diminuzione marginalmente significativa della vitalità è stata osservata alla dose di 5mM per AN3CA e RL95-2. Pertanto, la dose minima efficace di DCA per queste linee cellulari endometriali durante il periodo di trattamento è stata determinata tra 5mM e 10mM. Questa concentrazione di dose di DCA e il tempo di trattamento rientrano nell’intervallo di efficacia degli esperimenti in vitro pubblicati in precedenza [21]. È stato osservato un aumento statisticamente significativo della vitalità di HEC1A, HEC1B e KLE con l’aumento delle concentrazioni di DCA (p < 0,02). Come previsto, non è stata osservata alcuna differenza statisticamente significativa nella vitalità delle cellule epiteliali 293T in base all’intervallo di dosi di DCA e al periodo di trattamento (p = 0,27).
L’effetto del DCA sulla proliferazione delle cellule di cancro endometriale dipende dalla linea cellulare
Per determinare se la diminuzione della vitalità cellulare osservata con il trattamento con DCA fosse dovuta a un effetto di proliferazione cellulare, è stata eseguita una colorazione con BrdU / 7-AAD su diverse linee cellulari rappresentative e analizzata mediante citometria a flusso. Non sono state osservate differenze significative nella dinamica del ciclo cellulare o nella proliferazione nelle cellule epiteliali Ishikawa, HEC1B e 293T con il trattamento con DCA (Tabella 1). Nelle AN3CA, il trattamento con DCA ha aumentato la proliferazione, come indicato da un aumento significativo del numero di cellule nelle fasi S e G2/M e da un minor numero di cellule in G0/G1. Nelle cellule RL95-2, il trattamento con DCA ha ridotto significativamente la proporzione di cellule nelle fasi S e G2/M e ha aumentato le cellule nella fase G0/G1; ciò indica una diminuzione della proliferazione e un arresto del ciclo cellulare in uno stato senescente o quiescente.
| Linea cellulare / Trattamento | % Fase S | Stdev Fase S | % Fase G0 / G1 | Stdev G0/G1 | % G2 / M Fase | Stdev G2 / M Fase |
|---|---|---|---|---|---|---|
| 293T Non trattato | 60.6 | 0.6 | 33.2 | 1.7 | 6.2 | 1.25 |
| 293T DCA | 58.1 | 2.2 | 31.7 | 2.2 | 10.2 | 0.34 |
| valore p | 0.15 | 0.14 | 0.03 | |||
| Ishikawa Non trattato | 61.3 | 0.4 | 27.4 | 1.8 | 11.3 | 0.96 |
| Ishikawa DCA | 65.9 | 4.5 | 22.9 | 4.9 | 11.2 | 0.68 |
| valore p | 0.21 | 0.25 | 0.96 | |||
| HEC1B Non trattato | 37.4 | 2.7 | 41.5 | 0.8 | 21.2 | 1.93 |
| HEC1B DCA | 42.4 | 1.5 | 34.8 | 3.6 | 22.8 | 2.15 |
| valore p | 0.19 | 0.08 | 0.47 | |||
| AN3CA Non trattato | 43.5 | 0.6 | 50.6 | 2.0 | 5.9 | 1.80 |
IlDCA promuove l’apoptosi nelle cellule del cancro endometriale
Per determinare se la riduzione della vitalità cellulare dovuta al trattamento con DCA fosse dovuta all’apoptosi piuttosto che alla necrosi, è stata eseguita una colorazione delle cellule con Annexin-V-FITC e 7-AAD, analizzata mediante citometria a flusso. Sono stati osservati aumenti significativi dal 50% al 325% delle cellule apoptotiche precoci in AN3CA, Ishikawa, KLE, RL95-2 e SKUT1B (Figura 1B). Sono stati osservati aumenti significativi anche nelle cellule apoptotiche tardive di queste linee cellulari (Figura 1B). RL95-2 ha registrato l’aumento maggiore di cellule apoptotiche precoci, mentre KLE ha avuto l’aumento meno significativo. L’aumento della percentuale di cellule apoptotiche tardive in KLE non era statisticamente significativo. Non è stata osservata alcuna differenza nella percentuale di cellule apoptotiche precoci e tardive con il trattamento nelle cellule HEC1B, mentre le cellule HEC1A hanno registrato una diminuzione leggermente significativa delle cellule apoptotiche. La linea cellulare 293T non ha subito apoptosi con il trattamento con DCA e il leggero aumento della percentuale di cellule apoptotiche non è stato significativo (p=0,08).
Poiché il saggio dell’Annexin-V è utilizzato principalmente per rilevare l’apoptosi precoce, è stato eseguito un saggio TUNEL su diverse linee cellulari rappresentative di carcinoma endometriale per confermare qualitativamente la progressione verso l’apoptosi tardiva, visualizzando la frammentazione del DNA caspasi-dipendente. In accordo con il test quantitativo dell’Annexin-V, sono stati osservati aumenti delle cellule apoptotiche TUNEL-positive in AN3CA, Ishikawa e RL95-2 (Figura 1C). Nessuna differenza visiva nelle cellule TUNEL-positive è stata osservata nelle cellule HEC1B e 293T con il trattamento con DCA 10 mM.
Per determinare se il tasso di crescita influisce sulla sensibilità delle linee cellulari di carcinoma endometriale al trattamento con DCA, una responder apoptotica e una non-responder (rispettivamente Ishikawa e HEC1A) sono state coltivate in condizioni di privazione del siero con FBS allo 0,5%, che riporta le cellule alla fase G0 e ostacola la proliferazione. L’astinenza da siero non ha influenzato la percentuale di cellule apoptotiche precoci nelle cellule Ishikawa trattate rispetto alle normali condizioni di crescita. La percentuale di cellule apoptotiche precoci è aumentata dal 3,17% +/- 0,21% SD nelle cellule Ishikawa non trattate al 6,20% +/- 1,04% SD p=0,05 nelle cellule Ishikawa trattate, risultato simile a quello ottenuto in condizioni di crescita normali. La percentuale di cellule Ishikawa apoptotiche tardive è aumentata dall’1,07% +/- 0,15% SD nelle cellule non trattate al 3,57% +/- 0,49% SD p=0,02 nelle cellule trattate. La linea cellulare HEC1A non ha mostrato differenze significative nelle cellule apoptotiche precoci e tardive. La percentuale di cellule apoptotiche precoci di HEC1A è stata del 3,73% +/- 0,51% SD per le cellule non trattate e dell’1,93% +/- 0,60 SD p=0,07 per le cellule trattate. La percentuale di cellule apoptotiche tardive HEC1A era del 3,60% +/- 0,69% SD per le cellule non trattate e del 4,90% +/- 1,37 SD p=0,25 per quelle trattate.
L’apoptosi è mediata dalla diminuzione dei livelli di calcio intracellulare
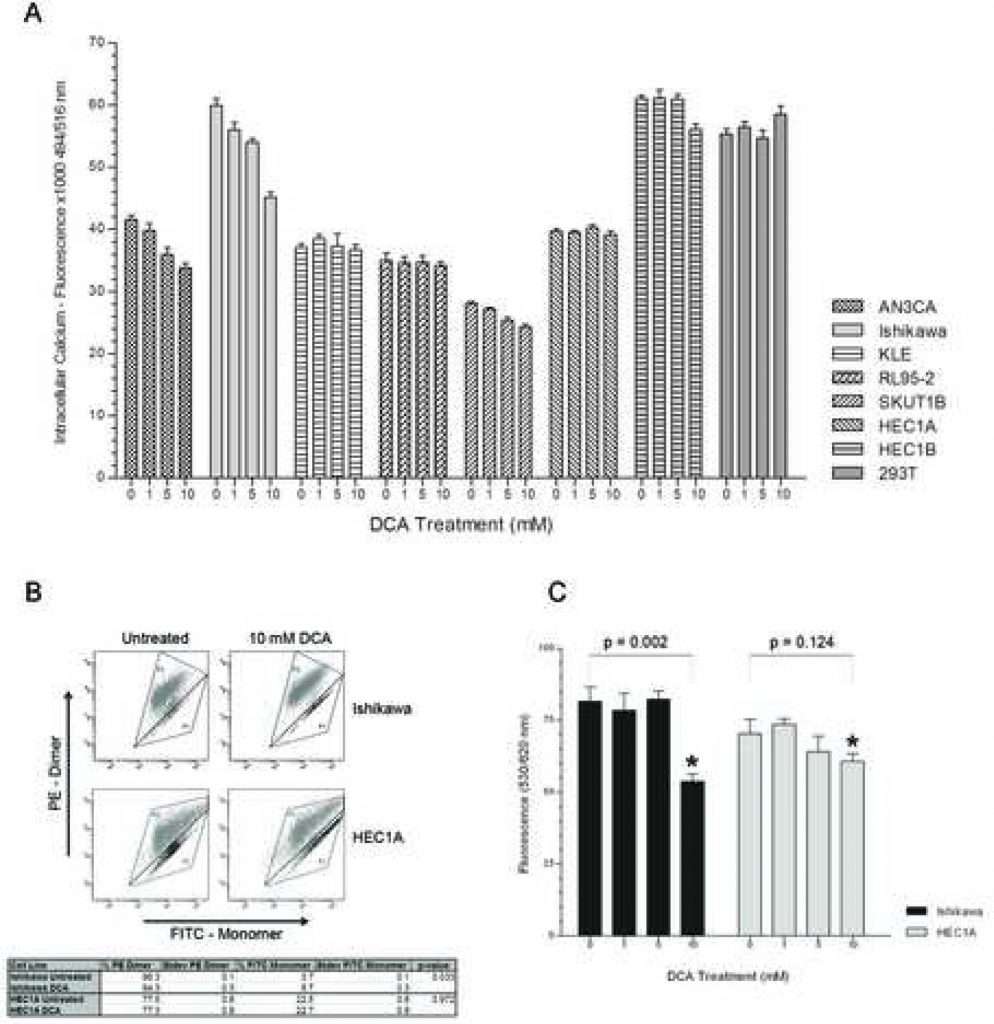
Per determinare se la via distale NFAT-Kv1.5 contribuisce alla promozione dell’apoptosi da parte del DCA, è stato eseguito un esperimento dose-risposta per valutare i livelli di Ca2+ intracellulare. AN3CA, Ishikawa e SKUTB hanno registrato livelli di calcio intracellulare decrescenti all’aumentare della dose di DCA (Figura 2A). La diminuzione del calcio intracellulare in queste linee cellulari ha raggiunto un livello significativo a una concentrazione di 5mM di DCA con p < 0,03 per AN3CA, Ishikawa e SKUT1B. La linea cellulare KLE, che ha mostrato la risposta apoptotica più lieve, ha registrato una diminuzione insignificante dei livelli di calcio con l’aumento della concentrazione di DCA. È interessante notare che non è stata rilevata alcuna differenza nei livelli di calcio intracellulare tra le dosi con RL95-2, che ha avuto la maggiore risposta apoptotica. HEC1A, che in precedenza non aveva mostrato alcuna risposta apoptotica, non presentava alcuna differenza nei livelli di calcio intracellulare a nessuna concentrazione di trattamento. Anche HEC1B non presenta differenze nei livelli di calcio tra i gruppi di trattamento con DCA 0, 1 e 5 mM e si osserva solo una leggera diminuzione con il trattamento a 10 mM. I livelli di calcio intracellulare di 293T non presentano differenze con l’aumento della concentrazione di DCA.
Iltrattamento con DCA riduce l’iper-polarizzazione della membrana mitocondriale nelle cellule di tumore dell’endometrio sottoposte ad apoptosi
Per valutare se il DCA abbia contribuito all’avvio dell’apoptosi attraverso un meccanismo regolato dai mitocondri, sono state misurate le MMP di una cellula responsiva all’apoptosi e di una non responsiva (rispettivamente Ishikawa e HEC1A), con e senza trattamento, mediante analisi FACS. Il reagente MitoCapture è un colorante cationico che, a seconda dell’entità del potenziale elettrico transmembrana mitocondriale, si accumula come monomero a emissione verde nel citoplasma o come dimero a emissione rossa nei mitocondri iperpolarizzati delle cellule tumorali [22, 23]. Il trattamento con DCA della linea cellulare Ishikawa ha ridotto la percentuale di cellule macchiate di rosso a 575 nm e aumentato la proporzione di cellule macchiate di verde a 525 nm, il che corrisponde alla sua risposta apoptotica al trattamento (Figura 2B). Il trattamento con DCA della linea cellulare HEC1A non ha influenzato la percentuale di cellule macchiate di rosso e verde. Inoltre, le cellule HEC1A non trattate presentavano una percentuale inferiore di cellule macchiate di rosso con membrane mitocondriali iperpolarizzate rispetto alle Ishikawa (77,5% +/- 0,6% SD vs. 96,3% +/- 0,1% SD, p < 0,01). Non vi è stata alcuna differenza nella MMP del controllo 293T non canceroso con il trattamento (dati non mostrati). La modulazione della MMP da parte del DCA è stata ulteriormente confermata utilizzando la colorazione TMRM in un esperimento dose-risposta. La MMP delle cellule Ishikawa è diminuita significativamente al dosaggio di 10 mM di DCA (Figura 2C). Non vi è stata alcuna differenza significativa nella MMP delle cellule HEC1A a qualsiasi concentrazione di trattamento.
IlDCA diminuisce l’espressione di Survivin
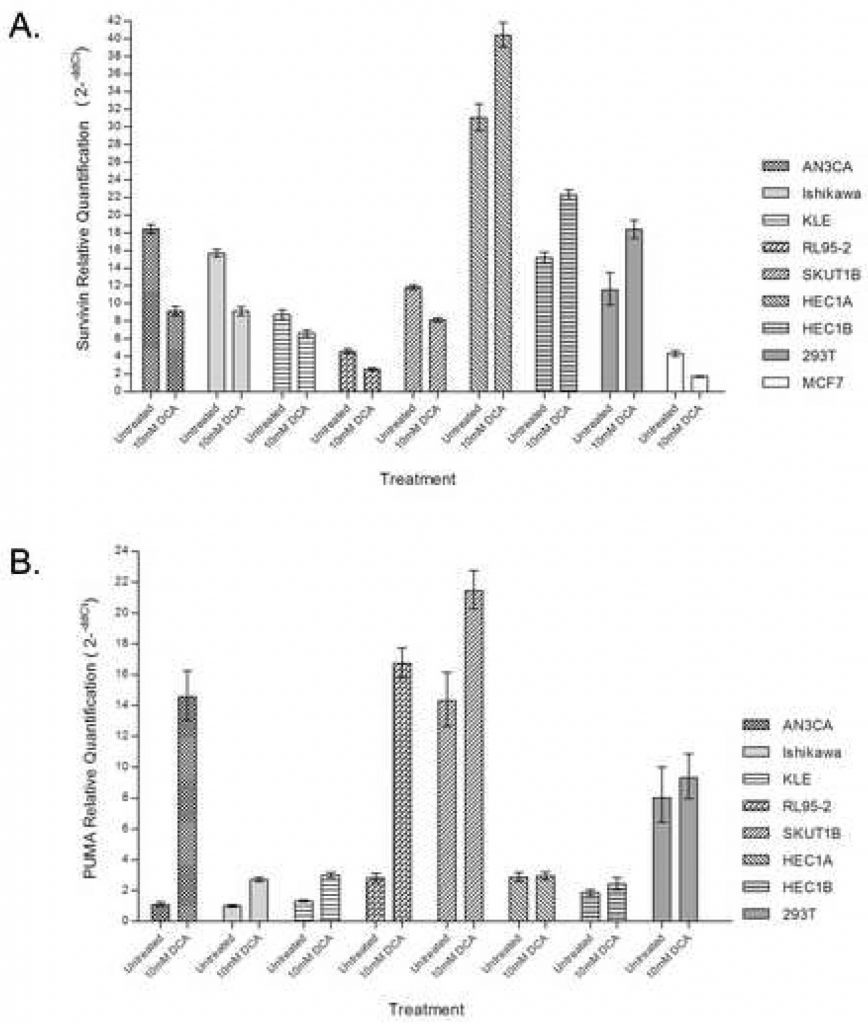
Per confermare ulteriormente il contributo della via prossimale regolata dai mitocondri alla risposta apoptotica, è stata utilizzata la PCR quantitativa in tempo reale per valutare l’espressione di Survivin con e senza somministrazione di DCA. Il trattamento delle linee cellulari di carcinoma endometriale con DCA ha determinato una diminuzione del 25%-50% dell’abbondanza di Survivin mRNA nelle cellule che hanno avuto una risposta apoptotica, tra cui AN3CA, Ishikawa, KLE, RL95-2 e SKUT1B (Figura 3A). RL95-2, che ha mostrato il maggiore aumento di cellule apoptotiche precoci con il trattamento, aveva anche la più bassa abbondanza di trascritto endogeno di Survivin. HEC1A e HEC1B, che in precedenza non avevano avuto una risposta apoptotica al DCA, hanno registrato un aumento del 20-30% dei trascritti di Survivin con il trattamento. HEC1A presentava la maggiore abbondanza di trascritti di Survivin endogeni. Anche il controllo 293T ha mostrato un aumento dell’abbondanza dei trascritti con il trattamento con DCA.
IlDCA aumenta l’espressione di PUMA
Per determinare il contributo della via PUMA nell’apoptosi indotta dal DCA, è stata utilizzata la PCR quantitativa in tempo reale per valutare l’abbondanza del trascritto di PUMA con e senza somministrazione di DCA. Il trattamento delle linee cellulari di cancro endometriale con DCA ha aumentato drasticamente l’abbondanza dell’mRNA di PUMA nelle linee cellulari che hanno avuto una risposta apoptotica, tra cui AN3CA, Ishikawa, KLE, RL95-2 e SKUT1B (Figura 3B). Il maggior grado di induzione di PUMA è stato osservato nelle cellule AN3CA e RL95-2, con aumenti rispettivamente di 14 e 6 volte. Le cellule HEC1A e HEC1B, che in precedenza non avevano avuto una risposta apoptotica al DCA, non hanno mostrato differenze nella quantità di trascritto PUMA con il trattamento. Nelle cellule 293T non è stata osservata alcuna differenza nella quantità di trascritto PUMA con il trattamento.
Discussione
In questo studio abbiamo dimostrato che la morte delle cellule di carcinoma endometriale indotta dal DCA è regolata da due meccanismi principali: la via regolata dai mitocondri e quella NFAT-Kv1.5. Inoltre, abbiamo dimostrato che il DCA riduce la vitalità delle cellule di cancro endometriale in modo dose-dipendente attraverso la promozione dell’apoptosi, mentre non ha alcun effetto sulle cellule 293T non cancerose. Infine, abbiamo dimostrato che il trattamento con DCA influenza la sopravvivenza delle cellule di cancro endometriale attraverso molteplici meccanismi molecolari, tra cui la regolazione del potenziale di membrana mitocondriale, i livelli di Ca2+ intracellulare, la perdita di espressione di Survivin e l’induzione di PUMA.
La risposta pro-apoptotica di AN3CA, Ishikawa e SKUT1B al DCA era correlata a una diminuzione dose-dipendente dei livelli di Ca2+ intracellulare, indicando il coinvolgimento del meccanismo NFAT-Kv1.5. In confronto, RL95-2 (che ha avuto la maggiore risposta apoptotica al DCA) e KLE (che ha avuto la risposta apoptotica più lieve) non hanno mostrato una differenza nei livelli di Ca2+ intracellulare a qualsiasi concentrazione di trattamento, suggerendo che la via del meccanismo NFAT-Kv1.5 non è coinvolta nel meccanismo apoptotico di queste linee cellulari. Invece, il trattamento con DCA ha arrestato le cellule RL95-2 nella fase G0/G1 del ciclo cellulare, un segno distintivo dell’attivazione di p53, ha fortemente indotto l’espressione di PUMA delle RL95-2 e ha ridotto l’espressione di Survivin, una proteina che svolge un ruolo critico nella regolazione del ciclo cellulare. [29].
La Survivin è un inibitore dell’apoptosi regolato a livello trascrizionale che, in risposta alle MMP interrotte, viene scaricato dai mitocondri al citoplasma, dove impedisce l’attivazione delle caspasi, inibisce l’apoptosi e promuove la progressione tumorale [26, 27]. Studi precedenti hanno dimostrato una correlazione positiva tra l’aumento dell’espressione di Survivin e il grado del tumore del carcinoma endometriale [27,28]. Abbiamo riscontrato che l’abbondanza del trascritto di Survivin è diminuita significativamente in tutte le linee cellulari di carcinoma endometriale che hanno avuto una risposta apoptotica al DCA. I nostri risultati indicano che la via regolata dai mitocondri contribuisce alla risposta apoptotica nelle linee cellulari di cancro endometriale sensibilizzate al DCA.
Il trascritto di PUMA è risultato significativamente aumentato in tutte le linee cellulari di cancro endometriale che hanno avuto una risposta apoptotica al DCA. Questo risultato può essere indicativo del contributo della via p53-PUMA con i meccanismi mitocondriali e dei canali ionici nell’apoptosi indotta dal DCA. L’aumentata espressione di PUMA contrasterebbe ulteriormente l’effetto pro-sopravvivenza di Bcl-2 sulla membrana mitocondriale nelle linee cellulari rispondenti, consentendo una maggiore traslocazione dei mediatori apoptotici dai mitocondri al citoplasma, promuovendo così una maggiore attivazione delle caspasi e l’apoptosi.
Due linee cellulari, HEC1A e HEC1B, entrambe altamente invasive con una maggiore resistenza ai farmaci e un grado tumorale più elevato [24] rispetto alle altre linee cellulari endometriali, sono risultate resistenti al DCA. Entrambe le linee cellulari hanno infatti mostrato un’aumentata espressione di Survivin, un’espressione di PUMA non influenzata dal trattamento con DCA e una minore percentuale di cellule con membrane mitocondriali iperpolarizzate. Questi risultati suggeriscono una minore dipendenza dalla glicolisi aerobica. Abbiamo valutato se le mutazioni nel dominio di legame del DCA della proteina PDK2 umana potessero spiegare le differenze nella risposta al trattamento con HEC1A e HEC1B. Tuttavia, dall’analisi della sequenza di tutte le linee cellulari non sono state riscontrate mutazioni nei due esoni che codificano il dominio putativo di legame del DCA (dati non mostrati).
In sintesi, il nostro studio dimostra che il dicloroacetato è efficace nel sensibilizzare all’apoptosi la maggior parte delle cellule di cancro endometriale a bassa o moderata invasività. I nostri dati collettivi suggeriscono che l’apoptosi è coerente con le vie mediate dai mitocondri e da NFAT-Kv1.5. Inoltre, i nostri dati suggeriscono che la via PUMA può essere coinvolta nella promozione apoptotica da parte del DCA. Le indagini future dovrebbero concentrarsi sull’esame di una gamma più ampia di tipi di cellule tumorali, oltre a determinare i meccanismi che conferiscono resistenza apoptotica al DCA. La via potenzialmente più informativa da prendere in considerazione è la caratterizzazione funzionale del ruolo della via PUMA nell’apoptosi indotta dal DCA.
Materiale supplementare
| Linea cellulare di cancro endometriale | Tipo di tumore | Grado di differenziazione | Invasività | stato p53 |
| AN3CA | Adenocarcinoma epiteliale | Indifferenziato (30, 31) | Moderato (25, 32) | Mutante (33) |
| KLE | Adenocarcinoma | Scarsamente differenziato (31, 34, 35) | Moderato (36) | Mutante (33) |
| Ishikawa | Adenocarcinoma epiteliale | Ben differenziato (25, 37) | Basso (25) | Mutante (33) |
| RL95-2 | Carcinoma epiteliale | Moderatamente differenziato (31, 35, 38) | Dipendente dal numero di passaggi (39) | Mutante (delezione di codone) (33) |
| SKUT1B | Leiomiosarcoma mesodermico | Ben differenziato | Moderato-alto (40) | n/a |
| HEC1A | Adenocarcinoma epiteliale | Moderatamente differenziato (31, 41) | Elevato (25, 42) | Mutante (33) |
| HEC1B | Adenocarcinoma epiteliale | Moderatamente differenziato (31, 41) | Alto (25, 42) | Mutante (33) |
Riconoscimenti
Desideriamo ringraziare il Dr. John Daley del Dana Farber HemNeo Flow Cytometry core facility per la formazione tecnica e la Dr.ssa Sabina Signoretti del Dana Farber – Harvard Cancer Center Pathology Core facility per il supporto immunoistochimico.
Sovvenzioni: Questo progetto è stato sostenuto dai numeri di sovvenzione CA082838 e CA101501 del National Institutes of Health.
RIFERIMENTI
1 Rose P. Carcinoma endometriale. New England Journal of Medicine. 1996;335(9):640-649. [PubMed] br>2 Ingram SS, Rosenman J, Heath R, Morgan TM, Moore D, Varia M. Il valore predittivo dei livelli del recettore del progesterone nel carcinoma endometriale. Int J Radiat Oncol Biol Phys. 1989;17(1):21-27. [PubMed] br>3 Randall ME, Filiaci VL, Muss H, et al. Studio randomizzato di fase III sull’irradiazione dell’intero addome rispetto alla chemioterapia con doxorubicina e cisplatino nel carcinoma endometriale avanzato: uno studio del Gynecologic Oncology Group. J Clin Oncol. 2006;24(1):36-44. [PubMed] br>4 Bonnet S, Archer SL, Allalunis-Turner J, et al. Un asse mitocondriale-K+ channel è soppresso nel cancro e la sua normalizzazione promuove l’apoptosi e inibisce la crescita del cancro. Cancer Cell. 2007;11(1):37-51. [PubMed] br>5 Chen Z, Lu W, Garcia-Prieto C, Huang P. The Warburg effect and its cancer therapeutic implications. J Bioenerg Biomembr. 2007 [PubMed] br>6 Kim JW, Dang CV. La dolcezza molecolare del cancro e l’effetto Warburg. Cancer Res. 2006;66(18):8927-8930. [PubMed] br>7 Chen LB. Potenziale di membrana mitocondriale nelle cellule viventi. Annu Rev Cell Biol. 1988;4:155-181. [PubMed] br>8 Heerdt BG, Houston MA, Augenlicht LH. Il potenziale di membrana mitocondriale intrinseco delle cellule di carcinoma del colon è legato alla probabilità di progressione del tumore. Cancer Res. 2005;65(21):9861-9867. [PubMed] br>9 Han J, Flemington C, Houghton AB, et al. L’espressione di bbc3, un gene pro-apoptotico BH3-only, è regolata da diversi segnali di morte e sopravvivenza cellulare. Proc Natl Acad Sci U S A. 2001;98(20):11318-11323. [Articolo libero da PMC] [PubMed] br>10 Nakano K, Vousden KH. PUMA, un nuovo gene pro-apoptotico, è indotto da p53. Mol Cell. 2001;7(3):683-694. [PubMed] br>11 Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induce la rapida apoptosi delle cellule di cancro del colon-retto. Mol Cell. 2001;7(3):673-682. [PubMed] br>12 Willis SN, Adams JM. Life in the balance: how BH3-only proteins induce apoptosis. Curr Opin Cell Biol. 2005;17(6):617-625. [Articolo libero da PMC] [PubMed] br>13 Jeffers JR, Parganas E, Lee Y, et al. Puma è un mediatore essenziale delle vie apoptotiche p53-dipendenti e indipendenti. Cancer Cell. 2003;4(4):321-328. [PubMed] br>14 Yu J, Wang Z, Kinzler KW, Vogelstein B, Zhang L. PUMA media la risposta apoptotica a p53 nelle cellule di cancro del colon-retto. Proc Natl Acad Sci U S A. 2003;100(4):1931-1936. [Articolo libero da PMC] [PubMed] br>15 Abemayor E, Kovachich GB, Haugaard N. Effetti del dicloroacetato sulla piruvato deidrogenasi cerebrale. J Neurochem. 1984;42(1):38-42. [PubMed] br>16 Lopaschuk GD, Saddik M. Il contributo relativo del glucosio e degli acidi grassi alla produzione di ATP nei cuori riperfusi dopo ischemia. Mol Cell Biochem. 1992;116(1–2):111–116. [PubMed] br>17 Stacpoole PW. La farmacologia del dicloroacetato. Metabolismo. 1989;38(11):1124–1144. [PubMed] br>18 Wong A, Cortopassi GA. Misurazione ad alta velocità del potenziale di membrana mitocondriale in una linea cellulare neurale utilizzando un lettore di piastre a fluorescenza. Biochem Biophys Res Commun. 2002;298(5):750-754. [PubMed] br>19 Yan J, Jiang J, Lim CA, Wu Q, Ng HH, Chin KC. BLIMP1 regola la crescita cellulare attraverso la repressione della trascrizione di p53. Proc Natl Acad Sci U S A. 2007;104(6):1841-1846. [Articolo libero da PMC] [PubMed] br>20 Bieche I, Parfait B, Tozlu S, Lidereau R, Vidaud M. Quantificazione dell’espressione genica del recettore degli androgeni nei tumori mammari sporadici mediante RT-PCR in tempo reale: evidenza che MYC è un gene AR-regolato. Carcinogenesi. 2001;22(9):1521–1526. [PubMed] br>21 Hassoun EA, Ray S. L’induzione dello stress ossidativo e della morte cellulare da parte dei sottoprodotti della disinfezione dell’acqua potabile, dicloroacetato e tricloroacetato, nelle cellule J774.A1. Biochimica e fisiologia comparate Tossicologia e farmacologia. 2003;135(2):119-128. [PubMed] br>22 Di Lisa F, Silverman HS, Hansford RG. Funzione mitocondriale e danno cellulare in singoli miociti cardiaci esposti ad anossia e riossigenazione. Transplant Proc. 1995;27(5):2829-2830. [PubMed] br>23 Piccoli C, Scrima R, Boffoli D, Capitanio N. Il controllo della citocromo c ossidasi sul sistema di fosforilazione ossidativa cellulare dipende dallo stato energetico mitocondriale. Biochem J. 2006;396(3):573-583. [Articolo libero da PMC] [PubMed] br>24 Liang Y, O’Driscoll L, McDonnell S, et al. Maggiore invasività in vitro e resistenza ai farmaci con modelli di espressione genica alterati in una linea cellulare di carcinoma polmonare umano dopo selezione a impulsi con farmaci antitumorali. Int J Cancer. 2004;111(4):484-493. [PubMed] br>25 Sillem M, Prifti S, Koumouridis A, et al. L’invasività corrisponde alla differenziazione piuttosto che alla secrezione di proteinasi in linee cellulari di cancro endometriale. European Journal of Gynaecologic Oncology. 1999;20(5–6):367–370. [PubMed] br>26 Dohi T, Okada K, Xia F, et al. Un complesso IAP-IAP inibisce l’apoptosi. J Biol Chem. 2004;279(33):34087–34090. [PubMed] br>27 Ambrosini G, Adida C, Altieri DC. Un nuovo gene anti-apoptosi, survivin, espresso nel cancro e nel linfoma. Nat Med. 1997;3(8):917-921. [PubMed] br>28 Erkanli S, Bolat F, Kayaselcuk F, Demirhan B, Kuscu E. COX-2 e survivin sono sovraespressi e positivamente correlati nel carcinoma endometriale. Gynecol Oncol. 2007;104(2):320-325. [PubMed] br>29 Li F, Ambrosini G, Chu EY, et al. Controllo dell’apoptosi e del checkpoint del fuso mitotico da parte della survivin. Nature. 1998;396(6711):580–584. [PubMed] br>30 Rice LW, Stone RL, Xu M, et al. Bersagli biologici per l’intervento terapeutico nell’adenocarcinoma endometriale endometrioide e nei tumori mulleriani misti maligni. Am J Obstet Gynecol. 2006;194(4):1119-1126. discussione 26-28. [PubMed] br>31 Nagamani M, Stuart CA. Legame specifico e attività di promozione della crescita dell’insulina in cellule di cancro endometriale in coltura. Am J Obstet Gynecol. 1998;179(1):6-12. [PubMed] br>32 Zhao Y, Yamashita T, Ishikawa M. Regolazione dell’invasione tumorale da parte del gene HOXB13 sovraespresso nel cancro endometriale umano. Oncol Rep. 2005;13(4):721-726. [PubMed] br>33 Yaginuma Y, Westphal H. Analisi del gene p53 in linee cellulari di carcinoma uterino umano. Cancer Res. 1991;51(24):6506-6509. [PubMed] br>34 Richardson GS, Dickersin GR, Atkins L, et al. KLE: una linea cellulare con recettore degli estrogeni difettoso derivata da un cancro endometriale indifferenziato. Gynecol Oncol. 1984;17(2):213-230. [PubMed] br>35 Carter CA, Parham GP. Lo stato di differenziazione influenza la risposta delle cellule di adenocarcinoma endometriale all’acido retinoico. Anticancer Research. 1997;17(3C):1973–1983. [PubMed] br>36 Yabushita H, Narumiya H, Hiratake K, et al. L’associazione del fattore di crescita trasformante-beta 1 con l’invasione miometriale dei carcinomi endometriali attraverso gli effetti sulla metalloproteinasi di matrice. J Obstet Gynaecol Res. 2000;26(3):163-170. [PubMed] br>37 Holinka CF, Hata H, Kuramoto H, et al. Risposte all’estradiolo in una linea cellulare di adenocarcinoma endometriale umano (Ishikawa) Journal of Steroid Biochemistry. 1986;24(1):85-89. [PubMed] br>38 Way DL, Grosso DS, Davis JR, et al. Caratterizzazione di un nuovo carcinoma endometriale umano (RL95-2) stabilito in coltura tissutale. In Vitro. 1983;19(3 parte 1):147-158. [PubMed] br>39 Sundareshan P, Hendrix MJ. Caratteristiche di crescita, morfologiche e invasive di passaggi precoci e tardivi di una linea cellulare di carcinoma endometriale umano (RL95-2) In Vitro Cell Dev Biol. 1992;28A(7-8):544-552. [PubMed] br>40 Colombatti A, Russo P, Cervi M, et al. Espressione differenziale di IRS-1 e IRS-2 in leiomiosarcomi uterini con fenotipi oncogenici distinti: Mancanza di correlazione con eventi di segnalazione a valle. Sarcoma. 2002;6(3):89-96. br>41 Kuramoto H, Tamura S, Notake Y. Stabilimento di una linea cellulare di adenocarcinoma endometriale umano in vitro. Am J Obstet Gynecol. 1972;114(8):1012–1019. br>42 Sieuwerts AM, Klijn JG, Foekens JA. Valutazione del potenziale invasivo di linee cellulari di tumori ginecologici umani con il saggio della camera di Boyden in vitro: influenze della capacità delle cellule di migrare attraverso la membrana del filtro. Clin Exp Metastasis. 1997;15(1):53-62.
Contenuti correlati: