Allison B. Haugrud, Yongxian Zhuang, Joseph D. Coppock, W. Keith Miskimins
Cancer Biology Research Center, Sanford Research, 2301 E. 60th St North, Sioux Falls, SD 57104, USA
e-mail: [email protected]
Ricevuto: 24 aprile 2014
Accettato: 5 settembre 2014
Pubblicato: 12 settembre 2014
Abstract
Il particolare metabolismo delle cellule del cancro al seno offre l’interesse di sfruttare questo fenomeno a livello terapeutico. La metformina, una promettente terapia per il cancro al seno, ha come bersaglio il complesso I della catena di trasporto degli elettroni e porta a un accumulo di specie reattive dell’ossigeno (ROS) che alla fine portano alla morte cellulare. L’inibizione del complesso I porta alla produzione di lattato, un sottoprodotto metabolico già altamente prodotto dalle cellule tumorali riprogrammate e associato a una prognosi infausta. Sebbene la metformina rimanga una promettente terapia antitumorale, abbiamo cercato un agente complementare in grado di aumentare gli effetti apoptotici della metformina, attenuando al contempo la produzione di lattato, con la possibilità di migliorare notevolmente l’efficacia. Il dicloroacetato (DCA) è un farmaco consolidato utilizzato nel trattamento dell’acidosi lattica che funziona attraverso l’inibizione della piruvato deidrogenasi chinasi (PDK) promuovendo il metabolismo mitocondriale. Il nostro scopo è stato quello di esaminare la sinergia e i meccanismi con cui questi due farmaci uccidono le cellule di cancro al seno. Le linee cellulari sono state sottoposte ai trattamenti indicati e analizzate per la morte cellulare e vari aspetti del metabolismo. La morte cellulare e la produzione di ROS sono state analizzate mediante citometria a flusso, analisi Western blot e metodi di conteggio delle cellule. Le immagini delle cellule sono state scattate con microscopia a contrasto di fase o microscopia confocale. Il metabolismo delle cellule è stato analizzato con l’analizzatore Seahorse XF24, il dosaggio del lattato e l’analisi del pH. Abbiamo dimostrato che quando il DCA e la metformina vengono utilizzati in combinazione, si verifica un’induzione sinergica dell’apoptosi delle cellule di cancro al seno. Il danno ossidativo indotto dalla metformina è potenziato dal DCA attraverso l’inibizione di PDK1, che riduce anche la produzione di lattato promossa dalla metformina. Dimostriamo che il DCA e la metformina si combinano per indurre sinergicamente l’apoptosi caspasi-dipendente che comporta un danno ossidativo con la contemporanea attenuazione della produzione di lattato promossa dalla metformina. Combinazioni innovative come metformina e DCA sono promettenti per l’espansione delle terapie del cancro al seno.
Parole chiave: Metformina; dicloroacetato; cancro al seno; lattato; apoptosi
© Springer Science+Business Media New York 2014
INTRODUZIONE
Il metabolismo del cancro sta diventando un’area promettente per lo sviluppo di nuovi approcci terapeutici. Rispetto alle cellule normali da cui derivano, le cellule tumorali sono metabolicamente riprogrammate e utilizzano preferenzialmente la glicolisi anche in condizioni di ossigeno sufficiente, un fenomeno noto come effetto Warburg [1]. Per compensare la perdita di ATP dovuta alla glicolisi preferenziale (rispetto alla progressione attraverso la fosforilazione ossidativa), le cellule tumorali registrano i geni che codificano i trasportatori di glucosio e gli enzimi glicolitici come la piruvato deidrogenasi chinasi (PDK) e la lattato deidrogenasi (LDH). L’elevato tasso di assorbimento del glucosio e l’alterazione del metabolismo non solo forniscono ATP e permettono alle cellule di sopravvivere in condizioni di ipossia, ma forniscono anche blocchi biosintetici, come intermedi e substrati per la produzione di aminoacidi, NADPH e ribosio-5-fosfato, essenziali per la sintesi di nucleotidi, proteine e membrane necessarie nelle cellule in rapida divisione. Ciò significa anche che il ciclo TCA mitocondriale genera una percentuale inferiore di ATP, permettendo al citrato di essere utilizzato nella biosintesi degli acidi grassi e dei lipidi per la produzione di nuove membrane [2]. Gran parte del lattato derivato dalla glicolisi viene assorbito dalle cellule circostanti con riciclo reciproco per sostenere la crescita del tumore e la resistenza ai meccanismi di morte cellulare apoptotica [2, 3]. Il lattato prodotto dai tumori può alterare il metabolismo delle cellule T e la presentazione dell’antigene da parte delle cellule dendritiche, con conseguente evasione immunitaria del tumore [4, 5]. Pertanto, gli elevati livelli di utilizzo del glucosio e la glicolisi offrono alle cellule tumorali numerosi vantaggi. Tuttavia, potrebbe anche essere possibile sfruttare il profilo metabolico unico del cancro a livello terapeutico. In questo studio abbiamo esaminato l’attività e l’interazione di due farmaci mirati al metabolismo, la metformina e il dicloroacetato (DCA), per determinare i loro effetti sulla crescita e sulla sopravvivenza delle cellule di cancro al seno.
La metformina cloridrato (1,1-dimetilbiguanide cloridrato) è un farmaco orale ampiamente utilizzato nel trattamento del diabete di tipo due. Alcuni studi hanno dimostrato che i pazienti diabetici che assumono metformina hanno una minore incidenza di cancro e di decessi correlati rispetto ai pazienti che non assumono metformina. In uno di questi studi, le pazienti diabetiche affette da cancro al seno che assumevano metformina avevano una risposta significativamente migliore alla chemioterapia neoadiuvante rispetto alle pazienti che non assumevano metformina [6-8]. Studi in vitro hanno concluso che la metformina inibisce la crescita di molti tipi di cellule tumorali, tra cui quelle del cancro al seno, del colon, della prostata, delle ovaie e dei gliomi [9-12]. È noto che la metformina attiva l’AMP-activated protein kinase (AMPK), che porta all’inibizione della sintesi proteica e della crescita cellulare [13]. Tuttavia, la sola attivazione dell’AMPK non è sufficiente a portare alla morte apoptotica delle cellule [14]. Alcuni studi hanno dimostrato che la metformina si accumula nei mitocondri e inibisce leggermente il complesso I della catena di trasporto degli elettroni, un evento che si verifica a monte dell’attivazione di AMPK [15-18]. Poiché il complesso I è inibito, l’ostacolo al passaggio degli elettroni porta alla produzione di superossido all’interno della matrice mitocondriale, danneggiando le proteine mitocondriali, i lipidi e gli acidi nucleici. Negli studi in cui è stato dimostrato che la metformina promuove la morte cellulare, l’apoptosi è la via principale [10, 12, 19]. In precedenza abbiamo dimostrato che la metformina induce la morte cellulare sia caspasi-dipendente che poli(ADP-ribosio) polimerasi (PARP)-dipendente nella maggior parte delle linee cellulari di cancro al seno, mentre non è citotossica per le cellule epiteliali mammarie non trasformate [20]. La morte cellulare dipendente dalla poli(ADP-ribosio) polimerasi è stata associata a importanti alterazioni della forma e della funzione mitocondriale, portando alla conclusione che il danno mitocondriale nelle cellule tumorali è un mediatore chiave della morte cellulare indotta dalla metformina. Sulla base di queste osservazioni, abbiamo ipotizzato che i composti che promuovono il metabolismo ossidativo mitocondriale aumentino il danno mitocondriale indotto dalla metformina e sinergizzino con quest’ultima nell’uccidere le cellule tumorali. Poiché il trattamento con metformina promuove anche la produzione di lattato [21], un composto di questo tipo dovrebbe idealmente combattere anche questo effetto.
Il dicloroacetato è anche un farmaco disponibile per via orale con una farmacocinetica ben studiata ed è stato testato per il trattamento dell’acidosi lattica (un potenziale effetto collaterale della metformina) e delle carenze mitocondriali [27]. Il dicloroacetato è un inibitore della PDK che fosforila la piruvato deidrogenasi (PDH), rendendola inattiva [23]. La piruvato deidrogenasi è l’enzima responsabile della trasformazione del piruvato in acetil-CoA per l’ingresso nel ciclo mitocondriale degli acidi tricarbossilici (TCA) e la fosforilazione ossidativa. Nelle cellule tumorali, l’attività della PDK è spesso elevata e agisce come un gatekeeper per ridurre il flusso di piruvato dal citoplasma al metabolismo mitocondriale. Si ritiene che questa sia una componente importante della riprogrammazione metabolica nelle cellule tumorali, che porta a una ridotta ossidazione del glucosio e alla produzione di lattato [24-26]. Inibendo la PDK, il DCA aumenta l’attività della PDH, consentendo al piruvato di entrare nel ciclo TCA anziché essere convertito in lattato e secreto [27].
In questo studio, abbiamo esaminato l’attività antitumorale e l’interazione di due farmaci mirati al metabolismo, la metformina e il DCA. Abbiamo dimostrato che il DCA aumenta la citotossicità della metformina sulle cellule di cancro al seno attraverso un meccanismo che coinvolge il danno ossidativo e contemporaneamente riduce la produzione di lattato da parte della metformina, fornendo potenzialmente un doppio vantaggio terapeutico.
I metodi
Prodotti chimici e reagenti
I seguenti prodotti chimici, reagenti e kit sono stati acquistati da Sigma-Aldrich, salvo diversa indicazione: metformina (1,1-dimetilbiguanide), dicloroacetato di sodio, soluzione di blu di tripan allo 0,
4 % di tripan blu, terreno di montaggio Vectashield per fluorescenza contenente 4,6 diamidino-2-fenilindolo (DAPI) (Vector Laboratories), kit per il dosaggio del lattato (Eton Biosciences), inibitore della caspasi OPH-109 (MP Biomedicals), Coomassie Brilliant Blue R250 (Bio-Rad Laboratories), paraformaldeide, SYTOX® Green (Life Technologies), Triton X-100 (Eastman) e inibitore PARP II INH2BP (Epigentek).
Coltura cellulare
Le linee cellulari di cancro al seno umano MCF7 e T47D e le cellule epiteliali mammarie umane MCF10A sono state acquistate da ATCC. La linea cellulare di carcinoma mammario di topo 66CL4 è stata fornita dal Dr. Fred Miller (Karmanos Cancer Institute, Detroit, MI). Una volta ricevute le linee cellulari, le cellule sono state immediatamente coltivate ed espanse per preparare stock di fiale congelate. Le cellule sono state fatte passare per non più di 2-3 mesi prima di stabilire nuove colture dalle fiale congelate di primo passaggio. Le linee cellulari sono state controllate di routine per la contaminazione da micoplasmi e sono state verificate dai laboratori IDEXX RADIL come esenti da micoplasmi. Le cellule sono state mantenute in terreno Dulbecco’s modified Eagle’s medium (DMEM) con 10 % di siero fetale bovino, 100 U/ml di penicillina e 100 µg/ml di streptomicina. Le cellule sono state incubate in un incubatorea CO2 umidificato a 37 °C.
Saggio di esclusione del blu di Trypan
Le linee cellulari MCF7 e T47D sono state piastrate su piatti da 35 mm. Le cellule sono state trattate con metformina e DCA alle concentrazioni indicate o con il veicolo. Dopo il periodo di tempo indicato, il terreno è stato raccolto per salvare le cellule morte galleggianti. Le cellule attaccate sono state lavate con soluzione salina tampone fosfato (PBS), che è stata riunita con il terreno raccolto. Le cellule sono state quindi raccolte mediante tripsinizzazione e aggiunte al pool. La soluzione di Trypan blue è stata utilizzata per colorare le cellule morte 1:1 e le cellule sono state contate con l’emitometro.
Western blotting
Le cellule sono state piastrate su piatti da 35 mm. Dopo il trattamento per il periodo di tempo indicato, le cellule sono state raccolte nel terreno di coltura per raccogliere le cellule vive e morte. Le cellule sono state pellettate, lavate con PBS e ripellicolate. Il pellet cellulare è stato quindi lisato con l’aggiunta di tampone per campioni 1× sodio dodecilsolfato (SDS) [2,5 mM Tris-HCl (pH 6,8), 2,5% SDS, 100 mM ditiotreitolo, 10% glicerolo, 0,025% blu di bromofenolo]. Quantità uguali di proteine sono state separate su un gel di SDS-poliacrilammide all’8,5%. Le proteine sono state trasferite su membrane Immobilon P (Millipore) con un’apparecchiatura Bio-Rad Trans-blot utilizzando un tampone di trasferimento [48 mM Tris-HCl, 39 mM glicina]. Le membrane sono state immerse nel 5% di latte non grasso secco in soluzione salina tamponata con Tris contenente Tween 20 (TBS-T) [10 mM Tris-HCl (pH 7,5), 150 mM NaCl, 0,1% Tween-20] con l’anticorpo indicato per 3 ore a temperatura ambiente o per 4 °C durante la notte. Dopo un accurato lavaggio con TBS-T, è stato applicato un anticorpo secondario appropriato coniugato alla perossidasi di rafano (HRP). La membrana è stata nuovamente lavata e le proteine sono state rilevate utilizzando il substrato chemiluminescente Super Signal West Pico (Pierce Biochemical). Per l’isolamento dei mitocondri, (Fig. 3d) le cellule MCF7 sono state coltivate al 95% di confluenza su piatti da 150 mm. Per isolare i mitocondri è stato utilizzato un kit di isolamento mitocondriale per cellule coltivate (Pierce), secondo il protocollo del produttore. Tutti i blot sono stati analizzati con un sistema di imaging UVP. Sono stati utilizzati i seguenti anticorpi: Piruvato deidrogenasi chinasi1 (Abcam), PARP (Cell Signaling), anti-4-idrossinonenale (Millipore), subunità del Complesso I NDUFB8 (Mitosciences), GAPDH (Ambion), PDH (Abcam) e fosfo-PDHE1 alfa (Calbiochem).
Microscopia confocale
Le cellule sono state disposte su vetrini di copertura in piatti da 35 mm e trattate come indicato. Le cellule sono state lavate in PBS, fissate con paraformaldeide al 4% per 10 minuti, montate in terreno Vectashield con DAPI su vetrini da microscopio standard e quindi osservate su un microscopio confocale Olympus FV1000 a 100×. La creazione di linee cellulari MCF7 che esprimono stabilmente pAcGFP1-Mito (Clontech; un plasmide che codifica proteine fluorescenti mirate ai mitocondri) è stata descritta in precedenza [20].
Saggio del lattato e analisi del pH
Le cellule MCF7 e 66CL4 in piastre da 35 mm sono state fatte crescere fino all’80% di confluenza e trattate come indicato in terreni privi di rosso fenolo e carbonato. Le cellule sono state trattate per 4-6 ore. Il terreno delle piastre è stato raccolto e la concentrazione di lattato è stata misurata utilizzando un kit per il dosaggio del lattato L disponibile in commercio (Eton Bioscience). Per la linea cellulare 66Cl4, le cellule sono state contate con l’emitometro per normalizzare i livelli di lattato. Per misurare il pH del terreno è stato utilizzato un pH-metro.
Saggio di formazione delle colonie
Le cellule MCF7 sono state piastrate a 500 cellule per piatto da 60 mm. Il giorno successivo le cellule sono state trattate come indicato. Dopo 12 giorni di incubazione, le cellule sono state lavate con PBS e fissate con etanolo al 70% per 5 minuti. Le colonie sono state quindi colorate con Coomassie Blue [40 % metanolo, 12 % acido acetico glaciale, 0,24 % Coomassie Blue], lavate, fotografate e quantificate utilizzando un sistema AlphaImager e il relativo software di analisi delle immagini (AlphaInnotech, Santa Clara, CA).
Citometria a flusso
Le cellule MCF7 sono state trattate come indicato. Le cellule sono state poi incubate con MitoSOX (5 µM) per 15 minuti, lavate, tripsinizzate e addizionate di 1 ml di DMEM al 10% di FBS. La rilevazione dei livelli di MitoSOX in un numero uguale di cellule per condizione di trattamento è stata determinata mediante citometria a flusso utilizzando un citometro Accuri C6.
Inibizione di PDK1 mediante un siRNA
On-TARGET Plus Smartpool siRNA mirati a PDK1 e siRNA non mirati sono stati acquistati da Thermo Scientific. le cellule 66CL4 (250.000/piatto) sono state piastrate su piatti da 35 mm. Il giorno successivo, le cellule sono state trasfettate con i siRNA utilizzando Dharmafect (Thermo Scientific) secondo il protocollo del produttore. La trasfezione è stata ripetuta il giorno successivo. Il giorno successivo, le cellule sono state lavate e trattate come indicato in DMEM senza carbonato con il 10 % di FBS. Dopo 4 ore, le cellule sono state contate con l’emitometro e il terreno è stato raccolto per misurare i livelli di pH e lattato. Le cellule sono state raccolte per l’analisi dei livelli di PDK1 mediante western blotting.
Saggio delle sostanze reattive dell’acido tiobarbiturico (TBARS)
Le cellule MCF7 sono state coltivate al 100% della confluenza su piatti da 150 mm. Le cellule sono state trattate come indicato per 24 ore con metformina (8 mM) o DCA (5 mM). Le cellule sono state raccolte in PBS, sonicate e i valori relativi delle proteine sono stati determinati con il saggio di Bradford. I lisati sono stati elaborati con il Quantichrom™ TBARS Assay Kit e letti su un lettore di piastre SpectraMax M5 (λ ex/em = 560 nm/585 nm).
SYTOX® green assay
Le cellule MCF7 e T47D sono state piastrate su piastre da 96 pozzetti e fatte crescere fino al 90% di confluenza. Le cellule sono state trattate come indicato, seguite dall’aggiunta del colorante per acidi nucleici SYTOX® Green (10 µM) e dall’incubazione per 20 minuti prima di essere lette su un lettore di piastre a fluorescenza a un λ ex/em = 485/535 nm con un cutoff di 515 nm. Le cellule sono state poi permeabilizzate con Triton X-100 (0,4%) per 30 minuti e una seconda lettura è stata acquisita per determinare il livello totale di colorazione del DNA, un surrogato del numero totale di cellule. I valori dell’indice di combinazione sono stati determinati utilizzando il software CalcuSyn.
Tasso di consumo di ossigeno
Il tasso di consumodi ossigeno(OCR) è stato misurato con un analizzatore Seahorse XF24 secondo le istruzioni del produttore (Seahorse Bioscience, North Billerica, MA, USA). Le cellule T47D sono state piastrate a 40.000 cellule/pozzetto in piastre XF24. Il giorno successivo, le cellule sono state lavate ed equilibrate con terreno privo di tampone per 30 minuti a 37 °C in un incubatoreprivo di CO2 prima di essere trasferite all’analizzatore XF24. È stata ottenuta una misurazione iniziale dell’OCR, seguita dall’aggiunta di DCA tramite porte di iniezione nei pozzetti a una concentrazione finale di 5 mM. L’OCR è stato misurato ogni 30 minuti.
Analisi statistica
Il confronto tra due gruppi è stato effettuato utilizzando il test t non accoppiato con correzione di Welch generato dal software Graph Pad Prism (La Jolla, CA). I valori di P inferiori o uguali a 0,05 sono stati considerati significativi.
Risultati
DCA e metformina inducono sinergicamente l’apoptosi nelle cellule di carcinoma mammario
In precedenza abbiamo dimostrato che la metformina induce due tipi distinti di morte cellulare dopo 3 giorni di trattamento nella maggior parte delle linee cellulari di carcinoma mammario, pur non essendo citotossica per le cellule epiteliali mammarie normali [20]. L’induzione della morte cellulare nelle cellule di carcinoma mammario era strettamente associata a un’alterazione della struttura mitocondriale, suggerendo che la metformina, noto inibitore del Complesso I della catena mitocondriale di trasporto degli elettroni, uccide le cellule tumorali alterando la funzione mitocondriale. Questo ci ha portato a ipotizzare che l’aumento del trasporto mitocondriale di elettroni possa aumentare la morte delle cellule tumorali indotta dalla metformina. Poiché il DCA promuove l’ossidazione mitocondriale del piruvato, abbiamo innanzitutto esaminato se il DCA potesse potenziare la citotossicità indotta dalla metformina nelle linee cellulari di cancro al seno.
Le cellule di cancro al seno MCF7 e T47D sono state trattate con metformina, DCA o la loro combinazione. La concentrazione di metformina utilizzata è stata di 8 mM, che rappresenta una dose fisiologicamente rilevante di metformina, come dimostrato dal lavoro di Owen et al. [17]. Come per il DCA, nel siero dei pazienti si possono raggiungere basse concentrazioni millimolari, quindi l’intervallo 0,5-5 mM è clinicamente rilevante [27-29] ed è stato utilizzato in questi esperimenti. Il numero di cellule vive e morte è stato determinato mediante il saggio di esclusione del blu di tripan (Fig. 1a). La morte cellulare è stata misurata nelle cellule MCF7 (Fig. 1a a sinistra) trattate per 2 giorni e nelle cellule T47D (Fig. 1a adestra) trattate per 4 giorni. In entrambe le linee cellulari, la metformina ha indotto la morte cellulare come osservato in precedenti esperimenti [20]. Tuttavia, la morte cellulare è stata significativamente aumentata dal trattamento concomitante con DCA e metformina, mentre il DCA da solo ha avuto effetti citotossici minimi (Fig. 1a). Quando le cellule epiteliali mammarie MCF10A non trasformate sono state sottoposte agli stessi trattamenti, la morte cellulare non è stata osservata, come mostrato nella Fig. 1b. Per approfondire l’osservazione della morte cellulare nelle cellule tumorali, sono state eseguite titolazioni della dose e saggi di citotossicità con SYTOX Green (Fig. 1c). La combinazione di farmaci è stata testata per la sinergia con il metodo di Chou [30] utilizzando il software CalcuSYN, che genera un indice di combinazione (CI) dei farmaci; la sinergia è indicata se il CI è inferiore a 1. Le cellule MCF7 (Fig. 1c a sinistra) avevano un CI di 0,00047 quando venivano trattate per due giorni con la combinazione di DCA e metformina. Le cellule T47D avevano un IC di 9,762e-006 dopo 4 giorni di trattamento (Fig. 1c a destra). Questi valori indicano una forte citotossicità sinergica con la combinazione di DCA e metformina in queste linee cellulari di cancro al seno.
Successivamente, gli effetti citotossici osservati di DCA (2,5 mM) e metformina (1 mM) sono stati convalidati utilizzando saggi di formazione di colonie. A queste concentrazioni, entrambi i farmaci da soli hanno ridotto le dimensioni delle colonie, ma non il loro numero (Fig. 1d, e). Tuttavia, è stata osservata una diminuzione significativa del numero di colonie quando i due farmaci sono stati combinati (Fig. 1e). Queste osservazioni dimostrano che il DCA potenzia la citotossicità indotta dalla metformina nelle cellule di carcinoma mammario rispetto al solo farmaco.
IlDCA promuove la fosforilazione ossidativa attraverso l’inibizione di PDK1 e attenua la produzione di lattato indotta dalla metformina
Per confermare che gli effetti osservati del DCA sono dovuti all’inibizione del bersaglio atteso, PDK1, dimostriamo che il DCA diminuisce effettivamente i livelli di PDH fosforilata rispetto al controllo nelle cellule MCF7 e MCF10A mediante western blot (Fig. 2a). La stimolazione della piruvato deidrogenasi in seguito al trattamento con DCA dovrebbe aumentare il metabolismo ossidativo del piruvato. Questo effetto è stato studiato utilizzando un analizzatore Seahorse XF24. Sono stati misurati gli OCR delle cellule MCF7 di controllo rispetto a quelle trattate con DCA (Fig. 2b). Le cellule trattate con dicloroacetato presentavano un OCR significativamente più elevato rispetto alle cellule di controllo.
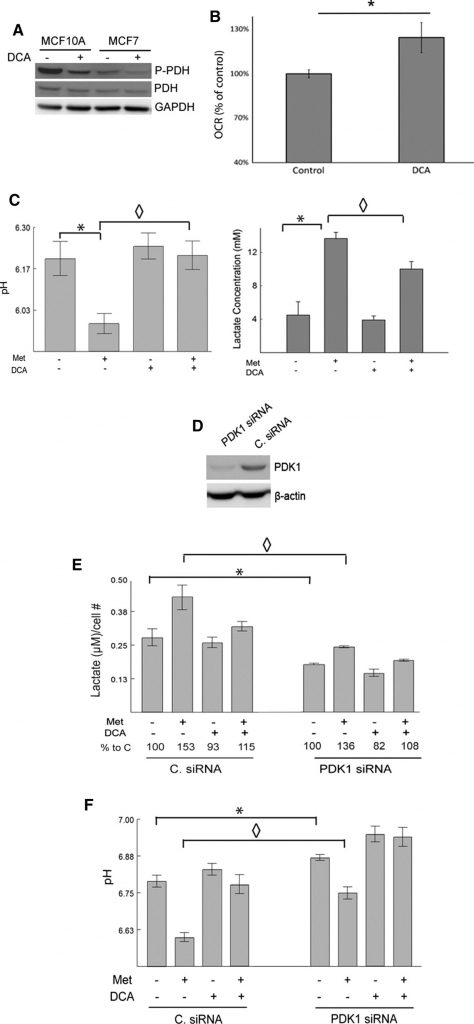
La metformina, in quanto blando inibitore del complesso I e potenziatore dell’attività di AMPK, dovrebbe stimolare i tassi glicolitici e aumentare la produzione di lattato [11,14,15,17]. Al contrario, il DCA dovrebbe reindirizzare il piruvato verso il metabolismo mitocondriale a scapito della produzione di lattato. Per esaminare questo aspetto, sono stati misurati i livelli di pH e di lattato nel terreno di coltura delle cellule MCF7 dopo il trattamento con metformina, DCA o entrambi. Le cellule trattate con metformina hanno prodotto livelli significativamente più elevati di lattato rispetto ai controlli non trattati. Il dicloroacetato da solo ha avuto effetti modesti, ma ha diminuito significativamente la produzione di lattato indotta dalla metformina. Il pH del terreno di coltura corrispondeva ai cambiamenti osservati nei livelli di lattato (Fig. 2c). In altre parole, la metformina ha ridotto significativamente il pH del terreno e questo effetto è stato ampiamente eliminato dal DCA.
Per dimostrare ulteriormente il ruolo della PDK nella produzione di lattato e nell’inibizione dell’ingresso del piruvato nel ciclo TCA, la PDK è stata abbattuta con un siRNA nelle cellule 66CL4. Questa linea cellulare deriva da un carcinoma mammario di topo e può essere utile per tradurre questi esperimenti in un contesto preclinico in studi futuri. Le cellule sono state trasfettate con un siRNA mirato a PDK1 (PDK1siRNA) o con un siRNA di controllo non mirato (C. siRNA). I livelli di PDK1 sono stati rilevati tramite western blot e le cellule trasfettate con PDK1 siRNA presentavano livelli notevolmente inferiori di PDK1 rispetto alle cellule trasfettate con C. siRNA (Fig. 2d). La repressione di PDK1 mediata da siRNA ha ridotto la produzione di lattato e aumentato il pH del mezzo, coerentemente con le funzioni note di PDK1 (Fig. 2e, f). La repressione di PDK1 ha ridotto efficacemente le variazioni di lattato e pH medio indotte dalla metformina. Il dicloroacetato ha ulteriormente ridotto il lattato e aumentato il pH nelle cellule trasfettate con PDK1 siRNA. La capacità del DCA di invertire gli effetti della metformina sul lattato e sul pH è stata in qualche modo attenuata quando PDK1 è stato eliminato, suggerendo ancora una volta che il DCA media i suoi effetti attraverso PDK1. In sintesi, si può concludere che la perdita di PDK1 mediante trasfezione con siRNA o inibizione con DCA previene la produzione di lattato indotta dalla metformina.
IlDCA in combinazione con la metformina aumenta il danno ossidativo e la conseguente morte cellulare caspasi-dipendente
La metformina inibisce il complesso I della catena di trasporto degli elettroni, impedendo il flusso di elettroni e portando alla produzione di superossido all’interno della matrice mitocondriale. Poiché il DCA promuove l’ingresso del piruvato nel ciclo TCA, abbiamo ipotizzato che il DCA possa aumentare la produzione di ROS come potenziale meccanismo della sua sinergia citotossica con la metformina. Il MitoSOX, un indicatore specifico della produzione di superossido mitocondriale, ha colorato le cellule che sono state analizzate mediante citometria a flusso. Dopo 1 ora di trattamento, né la metformina (linea rossa Fig. 3a pannello sinistro) né il DCA (non mostrato) hanno aumentato la produzione di superossido mitocondriale. Tuttavia, il trattamento combinato con DCA e metformina ha prodotto uno spostamento verso destra dell’intensità di fluorescenza, indicando un aumento della produzione di superossido. Dopo 3 ore di trattamento, le cellule trattate con metformina hanno iniziato a produrre livelli maggiori di superossido mitocondriale (linea rossa Fig. 3a pannello destro) rispetto alle cellule non trattate (linea nera). La combinazione di DCA e metformina ha mostrato livelli ancora maggiori di colorazione di superossido. Questi risultati indicano che la metformina interferisce con il trasporto di elettroni nel complesso I, portando a un aumento della produzione di superossido, e che il DCA potenzia l’effetto della metformina.
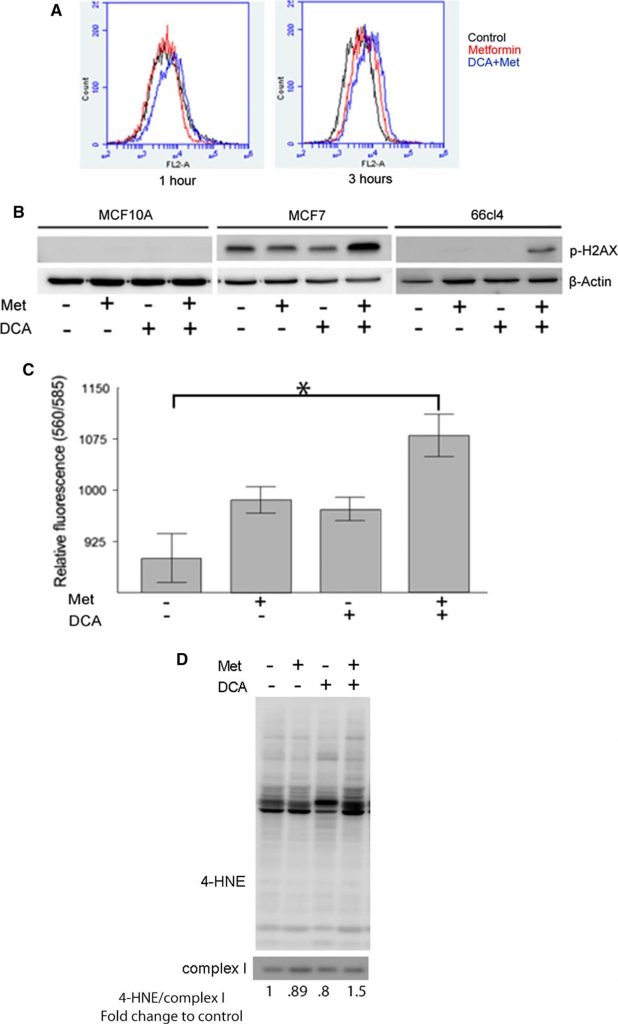
L’accumulo di superossido e di altri ROS ha il potenziale di danneggiare i componenti cellulari, tra cui proteine, membrane e DNA. Il danno ossidativo al DNA può portare a rotture del doppio filamento e alla fosforilazione dell’istone H2AX (p-H2AX), che è un utile marcatore surrogato di tale danno al DNA [31]. Pertanto, per stimare il danno al DNA associato alla produzione di ROS in risposta al trattamento con metformina e DCA, è possibile analizzare i livelli di H2AX fosforilato. L’epitelio mammario umano MCF10A e le linee cellulari di cancro al seno MCF7 e 66CL4 sono state analizzate mediante western blotting dopo il trattamento con DCA, metformina o la loro combinazione per 24 ore (Fig. 3b). Nelle cellule MCF10A non sono state osservate quantità rilevabili di p-H2AX con nessun trattamento. Nelle cellule MCF7, la p-H2AX era presente nelle cellule non trattate ed è stata notevolmente aumentata dalla combinazione di metformina e DCA, ma non da uno dei due farmaci da solo. Nelle cellule 66CL4, la p-H2AX è stata osservata solo nelle cellule trattate con DCA e metformina. Questi dati suggeriscono che DCA e metformina si combinano per aumentare il danno ossidativo al DNA.
Come secondo livello di evidenza, abbiamo misurato la perossidazione lipidica, che si verifica quando i radicali liberi danneggiano i lipidi nelle membrane cellulari. Per stimare l’entità dell’ossidazione lipidica in seguito al trattamento con DCA e metformina sono stati utilizzati due metodi distinti. In primo luogo, il test TBARS, che misura i sottoprodotti della perossidazione lipidica e viene utilizzato per valutare l’entità del danno ossidativo delle membrane cellulari causato dai ROS. Il TBARS è aumentato nelle cellule MCF7 trattate con metformina o DCA da soli, e l’aumento maggiore è stato osservato quando i due farmaci sono stati combinati (Fig. 3c). Il secondo metodo utilizzato per stimare l’ossidazione lipidica è stato il western blot degli addotti proteici del 4-idrossinonenolo (4-HNE), che derivano dall’ossidazione degli acidi grassi insaturi nelle membrane cellulari e formano addotti covalenti con le proteine, fornendo un’indicazione del danno ossidativo alle membrane cellulari. Gli estratti mitocondriali di cellule MCF7 trattate con DCA, metformina o entrambi per 24 ore sono stati analizzati per i livelli di addotti proteici 4-HNE (Fig. 3d). L’aumento dei livelli di addotti 4-HNE mitocondriali è stato osservato solo nelle cellule trattate con la combinazione di metformina e DCA. Ciò conferma i risultati precedenti, secondo cui la combinazione di questi farmaci provoca un aumento della produzione di ROS.
I nostri dati passati dimostrano che le cellule sensibili alla metformina sviluppano grandi vacuoli che, grazie all’imaging dei mitocondri legati alla GFP e alla microscopia elettronica, sono risultati essere mitocondri strutturalmente alterati. Questi mitocondri alterati sembrano essere specificamente legati a un percorso di morte cellulare PARP-dipendente che è ritardato nel tempo rispetto alla morte cellulare apoptotica [20]. Poiché il DCA sinergizza con la metformina nella morte cellulare, sono stati studiati gli effetti del DCA sulla struttura mitocondriale. È stata sviluppata una linea cellulare MCF7 stabile che esprime una proteina verde fluorescente mirata ai mitocondri (pAcGFP1-Mito). Le cellule sono state trattate per un giorno prima di essere sottoposte a imaging con contrasto di fase e microscopia confocale (Fig. 4). Nelle cellule trattate con la sola metformina sono stati osservati mitocondri di grandi dimensioni, come già visto in precedenza [20]. In netto contrasto, le cellule trattate con DCA sembravano avere mitocondri più piccoli e più numerosi. Il dicloroacetato ha anche impedito l’ingrandimento mitocondriale causato dalla metformina. Per contrasto di fase, i mitocondri ingranditi erano evidenti nelle cellule trattate con metformina, mentre erano assenti nel trattamento combinato. È stata esaminata anche una seconda linea cellulare sensibile alla metformina, la T47D. Anche in questo caso, sono stati osservati mitocondri ingranditi con il trattamento con metformina, mentre erano assenti nel trattamento combinato.
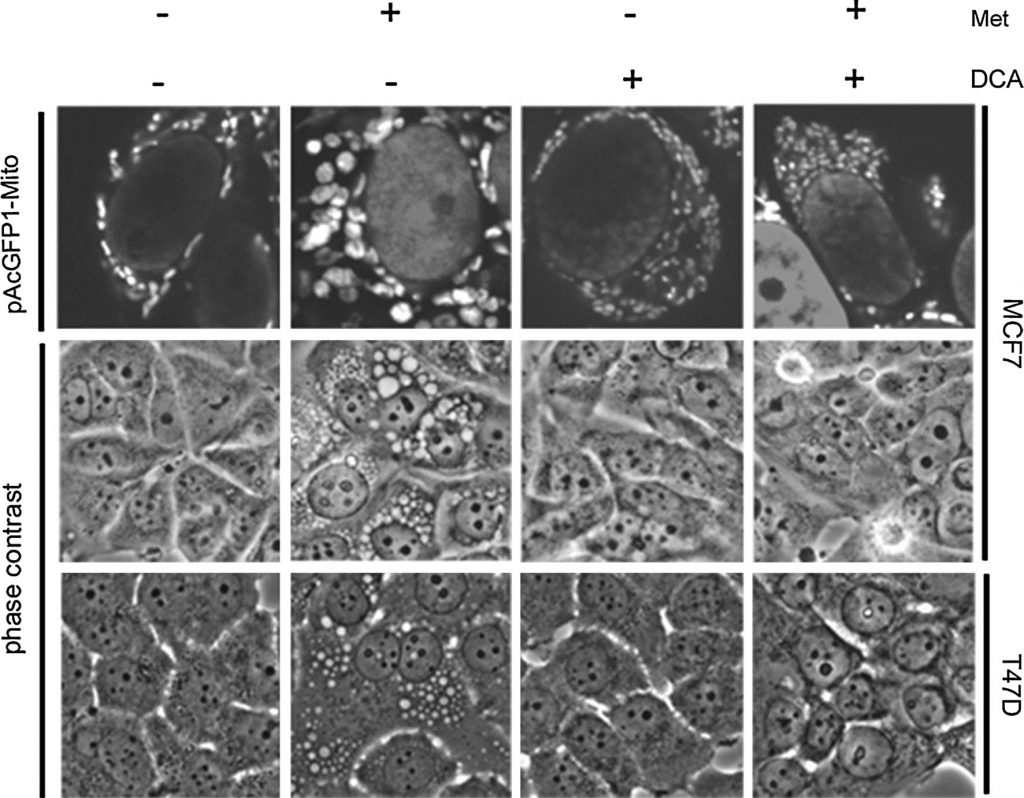
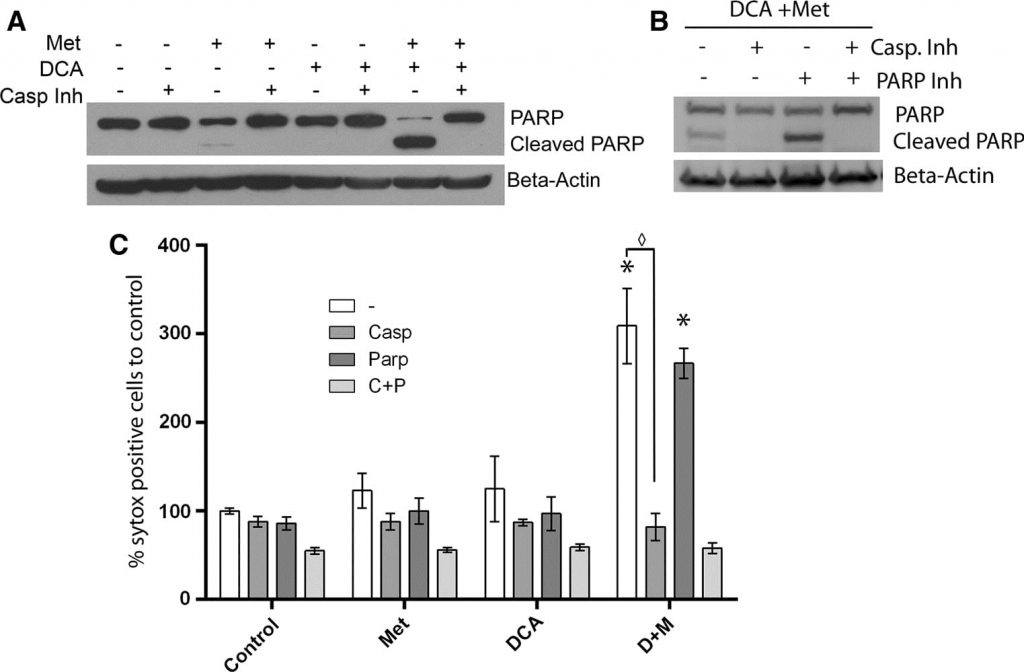
Nel nostro lavoro precedente, abbiamo scoperto che l’allargamento mitocondriale era correlato alla morte cellulare PARP-dipendente, che era ritardata nel tempo rispetto alla morte cellulare apoptotica. Ciò ha indotto a indagare sulle specificità del meccanismo di morte cellulare indotto dal DCA combinato con la metformina [20], poiché non si osserva alcun allargamento mitocondriale. Per caratterizzare la morte cellulare indotta nelle cellule co-trattate con metformina e DCA, le cellule MCF7 sono state trattate con un inibitore irreversibile delle caspasi ad ampio spettro (Q-Val-Asp-OPh) insieme a DCA, metformina o entrambi per un giorno. Il Western blotting (Fig. 5a) ha rivelato un forte aumento della PARP clivata e una diminuzione della PARP intatta nelle cellule trattate con DCA e metformina. L’aggiunta dell’inibitore della caspasi ha bloccato completamente la scissione di PARP. Un effetto simile era stato osservato in precedenza nelle cellule trattate con la sola metformina per 2,5 giorni [20]. Tuttavia, al termine di un giorno, la metformina da sola induce solo un leggero aumento della PARP clivata. Pertanto, questi dati indicano che l’apoptosi caspasi-dipendente è accelerata dall’aggiunta di DCA alla metformina in queste cellule di cancro al seno. Per verificare ulteriormente se la morte PARP-dipendente si verifica con il co-trattamento di DCA e metformina, è stato utilizzato anche un inibitore di PARP. È stato eseguito un saggio SYTOX Green (Fig. 5c) due giorni dopo il trattamento per quantificare la morte cellulare e scoprire se l’inibitore PARP avesse un effetto additivo rispetto all’inibitore delle caspasi. L’inibitore della PARP non ha avuto alcun effetto protettivo da solo né alcun effetto additivo insieme all’inibitore della caspasi. Un western blot ha confermato questo risultato (Fig. 5b) e ha rivelato che l’inibitore della PARP non ha impedito la scissione della PARP. Poiché l’allargamento mitocondriale è associato alla morte cellulare PARP-dipendente nelle cellule trattate con metformina, la mancanza di allargamento mitocondriale con l’aggiunta di DCA suggerisce che l’apoptosi avviene esclusivamente in modo caspasi-dipendente.
Discussione
La metformina è un farmaco utilizzato da tempo nel trattamento del diabete di tipo due. Allo stesso modo, il DCA è stato studiato in studi clinici per il trattamento dell’acidosi lattica e delle carenze mitocondriali. In precedenza è stato dimostrato che la metformina rallenta la proliferazione e promuove la morte cellulare di molte linee di cellule tumorali attraverso l’apoptosi [9, 10, 12, 32]. La metformina si concentra nei mitocondri dove inibisce il complesso I della catena di trasporto degli elettroni, causando un flusso anomalo di elettroni verso l’ossigeno e producendo superossido all’interno della matrice mitocondriale [22, Fig. 3]. La produzione di specie reattive dell’ossigeno è problematica per le cellule del cancro al seno, poiché molte di esse hanno una minore espressione di manganese superossido dismutasi (MnSOD), che normalmente elimina il superossido mitocondriale [33]. Sebbene gli effetti pro-apoptotici e di inibizione della crescita della metformina sul cancro siano degni di nota, la nostra osservazione che la metformina aumenta la glicolisi, come evidenziato dall’aumento della produzione di lattato in vitro [21], può limitare la sua efficacia come agente solitario. Poiché elevati livelli di lattato sono stati descritti come favorenti il tumore e sono associati a un esito clinico sfavorevole [26, 34-38], abbiamo cercato di attenuare la produzione di lattato indotta dalla metformina come mezzo per migliorare potenzialmente l’efficacia terapeutica. In questo studio abbiamo scoperto che il DCA contribuisce ad attenuare la produzione di lattato indotta dalla metformina e che questo avviene attraverso l’inibizione della PDK1.
I nostri dati dimostrano che il DCA e la metformina inducono sinergicamente l’apoptosi in misura maggiore rispetto al solo trattamento con metformina. I nostri dati dimostrano anche che il DCA dirotta il piruvato verso l’uso nella fosforilazione ossidativa, il che porta all’accumulo di ROS nel contesto dell’inibizione del complesso I da parte della metformina. Concludiamo che questo aumento della produzione di ROS porta a tassi di apoptosi più rapidi.
In un nostro precedente lavoro, abbiamo dimostrato che la metformina induce non solo la morte cellulare apoptotica caspasi-dipendente, ma anche una morte cellulare PARP-dipendente che si verifica successivamente e che è stata rivelata quando è stato utilizzato un inibitore pan-caspasi [20]. Questa forma di morte cellulare era associata a cambiamenti morfologici nei mitocondri. In questo studio, il DCA ha impedito i cambiamenti morfologici mitocondriali altrimenti indotti dalla metformina, mentre il co-trattamento ha portato a un tasso più rapido di morte cellulare. Inoltre, l’inibizione della pan-caspasi ha impedito quasi completamente la morte cellulare ed eliminato il clivaggio PARP associato al co-trattamento con metformina e DCA. L’insieme di questi dati indica che il meccanismo di morte cellulare guidato dal trattamento concomitante è l’apoptosi caspasi-dipendente e che l’apoptosi è accelerata rispetto alla sola metformina. È possibile che questo processo apoptotico sia troppo catastrofico e non richieda o lasci il tempo per l’allargamento mitocondriale associato alla morte cellulare PARP-dipendente della sola metformina.
Durante la preparazione di questo manoscritto, è stato pubblicato uno studio di Choi e Lim [39] che ha esplorato il co-trattamento di DCA e metformina nelle cellule tumorali. I nostri risultati confermano e ampliano in modo significativo i risultati di questo studio. Mentre lo studio di Choi e Lim si concentrava quasi esclusivamente sulle cellule HeLa, il nostro studio dimostra l’efficacia della combinazione di DCA e metformina in più linee cellulari di cancro al seno. È importante notare che questa combinazione di farmaci non uccide le cellule epiteliali mammarie non trasformate, MCF10A, nelle stesse condizioni in cui i farmaci uccidono le cellule tumorali. Abbiamo eseguito accurate titolazioni che dimostrano un forte sinergismo dei farmaci nell’indurre la morte cellulare nelle cellule di cancro al seno. Questi risultati indicano che il DCA sarà efficace a concentrazioni che possono essere raggiunte terapeuticamente nell’uomo [27, 29], e a livelli molto più bassi di quelli utilizzati negli esperimenti dello studio di Choi e Lim (10-20 mM) [39]. Idealmente, la dose massima di DCA utilizzata per trattare le cellule tumorali dovrebbe essere sufficientemente bassa da evitare effetti avversi come la neuropatia. In uno studio di Michelakis et al. i livelli sierici di DCA raggiunti erano di 0,5 mM in pazienti affetti da glioblastoma che assumevano 6,25 mg/kg per via orale due volte al giorno [28]. Dosi più elevate, come 25 mg/kg, sono associate a neuropatia dose-dipendente. Di conseguenza, le concentrazioni clinicamente rilevanti sono probabilmente nell’intervallo 0,5-5 mM.
Sia i risultati qui presentati che quelli di Choi e Lim [39] hanno dimostrato che la morte cellulare causata dalla combinazione di DCA e metformina è associata allo stress ossidativo. Abbiamo inoltre dimostrato che questo è associato alla produzione di superossido indotta dalla metformina da parte dei mitocondri. Il dicloroacetato ha ulteriormente aumentato la produzione di superossido mitocondriale in presenza di metformina, portando a danni ossidativi ai mitocondri, ai lipidi cellulari e al DNA. Nuove combinazioni come metformina e DCA sono promettenti per ampliare le terapie del cancro al seno.
Ringraziamenti
Questo articolo è stato finanziato da sovvenzioni della Susan G. Komen for the Cure (KG100497) e del National Cancer Institute dei National Institutes of Health (1R01CA180033). I nuclei di citometria a flusso e di imaging sono stati sostenuti dalle sovvenzioni COBRE P20GM103548 e P20GM103620 del National Institute of General Medical Sciences presso i National Institutes of Health.
Conflitto di interessi
Gli autori dichiarano di non avere conflitti di interesse.
Norme etiche
Gli autori dichiarano che gli esperimenti sono conformi alle leggi vigenti nel Paese in cui sono stati eseguiti.
RIFERIMENTI
1 Warburg O (1956) Sull’origine delle cellule cancerose. Science 123(80):309-314. doi:10.1016/S0306-9877(96)90136-X
2 DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB (2008) The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7:11-20. doi:10.1016/j.cmet.2007.10.002
3 Zamzami N, Kroemer G (2001) Il mitocondrio nell’apoptosi: come si apre il vaso di Pandora. Nat Rev Mol Cell Biol 2:67-71. doi:10.1038/35048073
4 Gottfried E, Kunz-Schughart LA, Ebner S et al (2006) Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 107:2013-2021. doi:10.1182/blood-2005-05-1795
5 Fischer K, Hoffmann P, Voelkl S et al (2007) Effetto inibitorio dell’acido lattico derivato da cellule tumorali sulle cellule T umane. Blood 109:3812-3819. doi:10.1182/blood-2006-07-035972
6 Jiralerspong S, Palla SL, Giordano SH et al (2009) Metformina e risposte patologiche complete alla chemioterapia neoadiuvante in pazienti diabetiche con cancro al seno. J Clin Oncol 27:3297-3302. doi:10.1200/JCO.2009.19.6410
7 Evans JMM, Donnelly LA, Emslie-Smith AM et al (2005) Metformin and reduced risk of cancer in diabetic patients. BMJ 330:1303-1304. doi:10.1136/bmj.38393.572188.EB
8 Libby G, Donnelly LA, Donnan PT et al (2009) New users of Metformin are at low risk of incident cancer. Diabetes Care 32:1620-1625. doi:10.2337/dc08-2175
9 Zakikhani M, Dowling R, Fantus IG et al (2006) Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res 66:10269-10273. doi:10.1158/0008-5472.CAN-06-1500
10 Isakovic A, Harhaji L, Stevanovic D et al (2007) Doppia azione antiglioma della metformina: arresto del ciclo cellulare e apoptosi dipendente dai mitocondri. Cell Mol Life Sci 64:1290-1302. doi:10.1007/s00018-007-7080-4
11 Hadad SM, Appleyard V, Thompson AM (2008) L’attivazione terapeutica di metformina/AMPK promuove il fenotipo angiogenico nel modello di tumore al seno MDA-MB-435 ERα negativo. Breast Cancer Res Treat 114:391. doi:10.1007/s10549-008-0016-3
12 Buzzai M, Jones RG, Amaravadi RK et al (2007) Il trattamento sistemico con il farmaco antidiabetico metformina ostacola selettivamente la crescita delle cellule tumorali con deficit di p53. Cancer Res 67:6745-6752. doi:10.1158/0008-5472.CAN-06-4447
13 Sarbassov DD, Ali SM, Sabatini DM (2005) Growing roles for the mTOR pathway. Curr Opin Cell Biol 17:596-603. doi:10.1016/j.ceb.2005.09.009
14 Zhuang Y, Miskimins WK (2008) L’arresto del ciclo cellulare nelle cellule di cancro al seno trattate con metformina comporta l’attivazione dell’AMPK, la downregulation della ciclina D1 e richiede p27Kip1 o p21Cip1. J Mol Signal 3:1-11. doi:10.1186/1750-2187-3-18
15 Hinke SA, Martens GA, Cai Y et al (2007) Il metil succinato antagonizza l’attivazione dell’AMPK indotta dalla biguanide e la morte delle beta-cellule pancreatiche attraverso il ripristino del trasferimento di elettroni mitocondriali. Br J Pharmacol 150:1031-1043. doi:10.1038/sj.bjp.0707189
16 Zou M-H, Kirkpatrick SS, Davis BJ et al (2004) Activation of the AMP-activated protein kinase by the anti-diabetic drug metformin in vivo. Ruolo delle specie reattive dell’azoto mitocondriale. J Biol Chem 279:43940-43951. doi:10.1074/jbc.M404421200
17 Owen MR, Doran E, Halestrap AP (2000) Evidenza che la metformina esercita i suoi effetti antidiabetici attraverso l’inibizione del complesso 1 della catena respiratoria mitocondriale. Biochem J 614:607-614. doi:10.1042/0264-6021:3480607
18 Carvalho C, Correia S, Santos MS et al (2008) Metformin promotes isolated liver mitochondria impairment. Mol Cell Biochem 308:75-83. doi:10.1007/s11010-007-9614-3
19d Kefas BA, Cai Y, Kerckhofs K et al (2004) Metformin-induced stimulation of AMP-activated protein kinase in beta-cells impairs their glucose responsiveness and can lead to apoptosis. Biochem Pharmacol 68:409-416. doi:10.1016/j.bcp.2004.04.003
20 Zhuang Y, Miskimins WK (2012) Metformin induces both caspase-dependent and poly(ADP-ribose) polymerase-dependent cell death in breast cancer cells. Mol Cancer Res 9:603-615. doi:10.1158/1541-7786.MCR-10-0343.Metformina
21 Bailey CJ, Wilcock C, Day C (1992) Effetto della metformina sul metabolismo del glucosio nel letto splancnico. Br J Pharmacol 105:1009-1013
22StacpoolePW, Lorenz AC, Thomas RG, Harman EM (1988) Il dicloroacetato nel trattamento dell’acidosi lattica. Ann Intern Med 108:58-63. doi:10.1056/NEJM198308183090702
23 Whitehouse BSUE, Cooper RH, Randle PJ (1974) Meccanismo di attivazione della piruvato deidrogenasi da parte del dicloroacetato e di altri acidi carbossilici alogenati. Biochem J 141:761-774. doi:10.1056/NEJM198308183090702
24 Hussien R, Brooks GA (2011) Mitochondrial and plasma membrane lactate transporter and lactate dehydrogenase isoform expression in breast cancer cell lines. Physiol Genomics 43:255-264. doi:10.1152/physiolgenomics.00177.2010
25 Kim J, Tchernyshyov I, Semenza GL, Dang CV (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3:177-185. doi:10.1016/j.cmet.2006.02.002
26 Wigfield SM, Winter SC, Giatromanolaki A et al (2008) PDK-1 regola la produzione di lattato nell’ipossia ed è associata a una prognosi sfavorevole nel cancro squamoso della testa e del collo. Br J Cancer 98:1975-1984. doi:10.1038/sj.bjc.6604356
27 Stacpoole PW, Kurtz TL, Han Z, Langaee T (2008) Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv Drug Deliv Rev 60:1478-1487. doi:10.1016/j.addr.2008.02.014
28 Michelakis ED, Sutendra G, Dromparis P et al (2010) Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2:31ra34. doi:10.1126/scitranslmed.3000677
29 Mori M, Yamagata T, Goto T et al (2004) Trattamento con dicloroacetato per la citopatia mitocondriale: effetti a lungo termine nel MELAS. Brain Develop 26:453-458. doi:10.1016/j.braindev.2003.12.009
30 Chou T (2010) Studi di combinazione di farmaci e quantificazione della loro sinergia con il metodo Chou-Talalay Studi di combinazione di farmaci e quantificazione della loro sinergia con il metodo Chou-Talalay. Cancer Res 70:440-446. doi:10.1158/0008-5472.CAN-09-1947
31 Mah L, Karagiannis TC (2010) γH2AX: un marcatore molecolare sensibile del danno e della riparazione del DNA. Leukemia 24:679-686. doi:10.1038/leu.2010.6
32 Alimova IN, Liu B, Fan Z et al (2009) La metformina inibisce la crescita delle cellule di cancro al seno, la formazione di colonie e induce l’arresto del ciclo cellulare in vitro. Cell Cycle 8:909-915. doi:7933 [pii]
33 Soini Y, Vakkala M, Kahlos K et al (2001) L’espressione di MnSOD è meno frequente nelle cellule tumorali dei carcinomi mammari invasivi rispetto ai carcinomi in situ o alle cellule epiteliali mammarie non neoplastiche. J Pathol 195:156-162. doi:10.1002/path.946
34 Walenta S, Wetterling M, Lehrke M et al (2000) High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers high lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical. Cancer Res 60:916-921
35 Leek R, Harris AL (2009) L’espressione della lattato deidrogenasi 5 nel tumore a cellule squamose della testa e del collo è correlata alla prognosi dopo radioterapia radicale o post-operatoria. Oncology 77:285-292. doi:10.1159/000259260
36 Isidoro A, Casado E, Redondo A et al (2005) I carcinomi mammari soddisfano l’ipotesi di Warburg e forniscono marcatori metabolici della prognosi del cancro. Carcinogenesi 26:2095-2104. doi:10.1093/carcin/bgi188
37 Isidoro A, Martínez M, Fernández PL et al (2004) L’alterazione del fenotipo bioenergetico dei mitocondri è un segno distintivo del cancro al seno, gastrico, polmonare ed esofageo. Biochem J 378:17-20. doi:10.1042/BJ20031541
38 Walenta S, Schroeder T (2004) Lactate in solid malignant tumors: potential basis of a metabolic classification in clinical oncology. Curr Med Chem 11:2195-2204
39 Choi YW, Lim IK (2014) Sensibilizzazione della citotossicità della metformina da parte del dicloroacetato attraverso la riprogrammazione del metabolismo del glucosio nelle cellule tumorali. Cancer Lett 346:300-308. doi:10.1016/j.canlet.2014.01.015
Contenuti correlati: