Jingtao Tong, Ganfeng Xie, Jinxia He, Jianjun Li, Feng Pan e Houjie Liang
Dipartimento di Oncologia, Ospedale Sud-Ovest, Terza Università Medica Militare, 29 Gaotanyan Street, Chongqing 400038, Cina
La corrispondenza va indirizzata a Houjie Liang, [email protected] Ricevuto il 27 maggio 2010; rivisto il 29 dicembre 2010; accettato il 13 gennaio 2011
Editore accademico: Miguel A. Andrade
Copyright © 2011 Jingtao Tong et al. Questo è un articolo ad accesso libero distribuito secondo la Creative Commons Attribution License, che ne consente l’uso, la distribuzione e la riproduzione illimitata su qualsiasi supporto a condizione che l’opera originale sia adeguatamente citata.
Il dicloroacetato (DCA), un inibitore della piruvato deidrogenasi chinasi (PDK), è stato recentemente dimostrato come un promettente agente antineoplastico non tossico che promuove l’apoptosi delle cellule tumorali. Nel presente studio, ci siamo posti l’obiettivo di indagare l’effetto antitumorale del DCA combinato con il 5-fluorouracile (5-FU) sulle cellule di cancro del colon-retto (CRC). Quattro linee cellulari umane di CRC sono state trattate con DCA o 5-FU, o con una combinazione di DCA e 5-FU. La vitalità cellulare è stata determinata con il saggio del 3-(4,5-dimetiltiazol-2-il)- 2,5-difeniltetrazolio bromuro. L’interazione tra DCA e 5-FU è stata valutata con il principio dell’effetto mediano. L’immunocitochimica con bromodeossiuridina (BrdU) è stata effettuata per determinare la proliferazione delle cellule CRC. Il ciclo cellulare e l’apoptosi sono stati misurati mediante citometria a flusso e l’espressione di molecole correlate all’apoptosi è stata valutata mediante western blot. I nostri risultati hanno dimostrato che il DCA ha inibito la vitalità delle cellule CRC e ha avuto un effetto antiproliferativo sinergico in combinazione con il 5-FU. Inoltre, rispetto al 5-FU da solo, l’apoptosi delle cellule CRC trattate con DCA e 5-FU è stata potenziata e dimostrata con i cambiamenti delle proteine Bcl-2, Bax e della caspasi-3. I nostri risultati suggeriscono che il DCA ha un effetto antitumorale sinergico con il 5-FU sulle linee cellulari CRC in vitro.
1. Introduzione
Il cancro del colon-retto è uno dei tumori maligni più comuni al mondo [1]. Oltre alla chirurgia, il trattamento dei pazienti affetti da CRC si basa principalmente sulla chemioterapia, soprattutto per i pazienti con CRC avanzato. Tra gli agenti chemioterapici per il CRC, il 5-fluorouracile (5-FU), che è un agente chemioterapico classico, è stato il regime di prima linea per il trattamento del CRC per diversi decenni [2, 3]. Tuttavia, il 5-FU da solo è scarsamente selettivo nei confronti del tumore e altamente tossico per il midollo osseo, il tratto gastrointestinale e la pelle quando viene utilizzato alla dose terapeutica [4].
L’anormalità metabolica è una delle caratteristiche fondamentali del cancro [5]. Già negli anni ’20, Otto Warburg osservò che le cellule tumorali generalmente utilizzano la glicolisi piuttosto che la fosforilazione ossidativa per ottenere energia [6]. Pertanto, il passaggio metabolico alla respirazione anaerobica attraverso la glicolisi dal piruvato, piuttosto che la conversione del piruvato in acetil-CoA per azione della piruvato deidrogenasi (PDH) nel metabolismo aerobico del glucosio, diventa un fenotipo preferenziale del progresso del cancro. La PDH può essere inattivata dalla piruvato deidrogenasi chinasi (PDK) in molti fenotipi glicolitici, compreso il cancro, mentre l’inibizione della PDK sposta il metabolismo verso l’ossidazione aerobica, che si dimostra svantaggiosa per la crescita tumorale [7].
Il dicloroacetato (DCA) è un prototipo di inibitore della PDK mitocondriale. Bloccando l’enzima, il DCA riduce la produzione di lattato spostando il metabolismo del piruvato dalla glicolisi all’ossidazione nei mitocondri. Questa proprietà ha portato alla sperimentazione del DCA per il trattamento dei disturbi da accumulo di acido lattico [8]. Recentemente, alcuni studi hanno dimostrato che il DCA sopprime la crescita tumorale attraverso l’inibizione della PDK [9-11]. Michelakis e colleghi hanno scoperto che il DCA ripristina la funzione mitocondriale, ripristinando così l’apoptosi, uccidendo le cellule tumorali in vitro e riducendo i tumori nei ratti [12].
La chemioterapia combinata è stata ampiamente utilizzata. il 5-FU viene solitamente combinato con altri agenti antineoplastici e con le radiazioni per potenziare il suo effetto antitumorale. L’insufficienza clinica sembra essere causata dalla resistenza del 5-FU e dai gravi effetti collaterali. La forte e selettiva induzione dell’apoptosi suggerisce che l’inibitore PDK DCA possa potenziare l’effetto inibitorio dei farmaci antitumorali, superando così l’efficacia del trattamento attuale. In questa sede, ci siamo proposti di esaminare gli effetti antitumorali combinati del DCA con il 5-FU sulle cellule del CRC, nella speranza di valutare un regime relativamente efficace e sicuro potenziato per il trattamento del CRC.
2. Materiale e metodi
2.1. Cellule e componenti. Le linee cellulari umane di cancro del colon LS174T, LoVo, SW620 e HT29 sono state acquistate dall’American Type Culture Collection (Manassas, VA, USA). I reagenti per la coltura cellulare sono stati acquistati da Gibco-Invitrogen (Carlsbad, CA, USA). Le linee cellulari sono state mantenute in terreno Dulbecco’s Modified Eagle’s medium o in terreno Leibovitz L-15 contenente 10% di siero fetale bovino (FBS), 100 U/mL di penicillina e 100 μg/mL di streptomicina in un incubatore umidificato a 37◦C e 5% di CO2. 5-FU e DCA sono stati acquistati da Sigma-Aldrich Co. Ltd. (St. Louis, MO, USA), disciolti in acqua deionizzata per ottenere una soluzione di lavoro 1 mol/L, sterilizzati in filtro e successivamente diluiti nel terreno di crescita per il trattamento.
2.2. Saggio di vitalità cellulare. La vitalità cellulare è stata determinata con il saggio del 3-(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazolio bromuro (MTT) (Sigma-Aldrich). Le cellule sono state seminate in piastre da 96 pozzetti (5 × 104 cellule per pozzetto) e incubate in condizioni di crescita standard per una notte dal 60% al 70% di confluenza. Le cellule sono state poi trattate con DCA da solo (concentrazione finale 0-90 mM) o con DCA combinato con 5-FU (5-200 μM). Dopo un trattamento di 48 ore, le cellule sono state incubate a 37◦Cper altre 4 ore con MTT (20 μLper pozzetto) e l’assorbanza a 490 nm è stata misurata in un lettore di piastre BioRad Model 550 (Hercules, CA, USA).
2.3. Analisi dell’interazione tra farmaci. L’interazione tra DCA e 5-FU è stata analizzata utilizzando il principio dell’effetto mediano descritto da Chou e Talalay [13, 14]. Il programma consente di calcolare gli indici di combinazione (CI) che, se minori di 1, uguali a 1 o maggiori di 1, indicano rispettivamente sinergismo, additività o antagonismo tra due farmaci. Gli IC sono stati calcolati con (Dx)1 e (Dx)2, dove (Dx)1 e (Dx)2 sono le concentrazioni di DCA da solo o di 5-FU da solo, che danno un’inibizione della crescita dell’x%, e (D)1 e (D)2 sono le concentrazioni di farmaci in combinazione che inibiscono la crescita cellulare anche dell ‘x%.
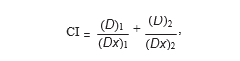
(Dx)1 e (Dx)2 sono stati calcolati con l’equazione mediana-effetto:
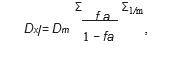
dove Dm è la dose mediana-effetta, fa è la frazione affetta e m rappresenta la pendenza del grafico mediana-effetta.
2.4. Saggi di proliferazione cellulare. L’immunocitochimica è stata effettuata con bromodeossiuridina (BrdU) (BD Bioscience, San Jose, CA, USA) in vitro. Le cellule sono state propagate su vetrini coprioggetto in piastre a 12 pozzetti in condizioni di crescita standard. Dopo 24 ore, sono state aggiunte varie concentrazioni di DCA, 5-FU o una combinazione di due farmaci. Le cellule sono state messe in astinenza da siero per 12 ore in terreni di crescita contenenti lo 0,5% di FBS per resettare il ciclo cellulare alla fase G0, quindi sono state pulsate per 2 ore con 10 μmol/L diBrdU in terreni di crescita. Successivamente, le cellule sono state fissate, lavate e colorate secondo le istruzioni del produttore.
L’analisi del ciclo cellulare è stata determinata indirettamente utilizzando la colorazione con ioduro di propidio (PI, BD Bioscience) mediante citometria a flusso (FACScan, Becton Dickinson, San Jose, CA, USA). Le cellule sono state seminate in piastre a 6 pozzetti e coltivate con o senza 10 mM DCA o 20 μM5-FU. Dopo un’incubazione di 48 ore, le cellule sono state raccolte con 0,25% Trypsin-EDTA (Invitrogen, Carlsbad, CA, USA). Quindi, le cellule sono state fissate con alcol al 70% per 24 ore a 4◦Ce lavate due volte con soluzione salina fosfato buffer (PBS). È stata aggiunta la RNAasi (100 μL; 1 mg/mL) (BD Bioscience) e le cellule sono state incubate in un bagno d’acqua a 37◦Cper 30 minuti. Dopo la colorazione con 200 μL diPI (50 μg/mL), le cellule sono state tenute a 4◦Cper 30 minuti. Infine, le cellule sono state analizzate mediante citometria a flusso.
2.5. Saggio di apoptosi. L’apoptosi è stata rilevata mediante citometria a flusso con annexina-V-FITC (BD Bioscience) e PI. Le cellule sono state seminate in piastre a 6 pozzetti. Dopo un’incubazione di 48 ore con o senza farmaci, le cellule sono state lavate e risospese in 0,5 mL di PBS buffer. Dopo la colorazione con annexin-V-FITC e PI, le cellule sono state analizzate mediante citometria a flusso in tre esperimenti indipendenti.
2.6. Western Blot. Le cellule sono state raccolte e le proteine totali sono state estratte con il buffer RIPA contenente inibitori delle proteasi. Le proteine totali (50 μg) sono state sottoposte a SDS/PAGE al 10% o al 12% e le proteine risolte sono state trasferite elettroforeticamente su membrane PVDF (Millipore, Bedford, MA, USA). Le membrane sono state bloccate per 2 ore con il 5% di latte non grasso in TBS buffer contenente lo 0,05% di Tween- 20 (TBST) a 4◦C. Le membrane sono state poi incubate con anticorpi anti Bax, Bcl-2, Caspasi-3 e GADPH per una notte a 4◦C. Dopo il lavaggio in TBST, le membrane sono state incubate con i rispettivi anticorpi secondari per 1 ora.
Le membrane sono state poi incubate con SuperSignal West Femto Maximum Sensitivity Substrate (Pierce, Rockford, IL, USA) per 1 minuto e sottoposte a imaging con un sistema Gel Doc XR (Bio-Rad). Tutti gli anticorpi sono stati acquistati da Santa Cruz Biotechnology (Santa Cruz, CA, USA).
2.7. Statistiche. Tutti i dati sono espressi come media della deviazione standard (SD). L’analisi statistica è stata eseguita con il software SPSS 13.0 (SPSS, Chicago, IL, USA). Le differenze tra i due gruppi sono state determinate mediante il t-test a campioni appaiati o il t-test a campioni indipendenti (a due code), come indicato. Le differenze tra i gruppi sono state analizzate mediante analisi della varianza (ANOVA) a una via. P < .05 è stato considerato statisticamente significativo.
3. Risultati
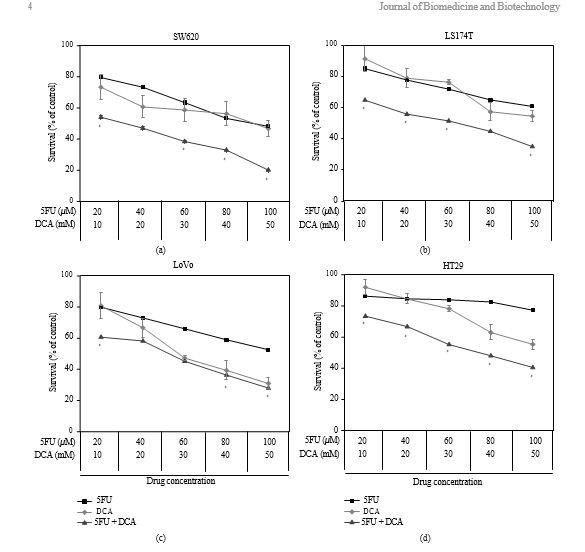
3.1. Vitalità delle cellule CRC trattate con DCA da solo o in combinazione con 5-FU. Per determinare l’effetto del DCA sulle cellule CRC, le cellule sono state esposte al DCA (0-90 mM) per 48 ore. I risultati hanno mostrato che l’effetto inibitorio era dose-dipendente. Come mostrato nella Figura 1, l’inibizione della vitalità delle linee cellulari tumorali trattate con 50 mM DCA è stata
come segue: SW620 (46,73% ± 5 ,21%), LoVo (30,94% ± 3,57%), LS174t (54,59% ± 3 ,93%) e HT29 (55,31% ± 3,35%). Abbiamo trattato le cellule con 5-FU (20-100 μM) e abbiamo riscontrato che la vitalità di SW620, LoVo, LS174t è stata inibita in modo significativo ad eccezione di HT29, la cui inibizione della vitalità non era evidente con 80 μM di5-FU. Quando sono state trattate con questi due farmaci contemporaneamente, la vitalità delle cellule CRC summenzionate è diminuita significativamente rispetto al DCA o al 5-FU da soli (i valori IC50 sono riportati nella Tabella 1).
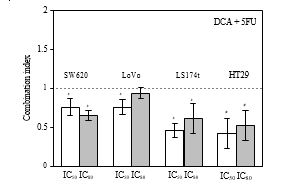
3.2. Effetti sinergici del DCA combinato con il 5-FU nelle linee cellulari CRC. La vitalità delle cellule CRC è diminuita in presenza di 5-FU e DCA. Il DCA ha potenziato gli effetti inibitori del 5-FU sulle cellule CRC e l’influenza del DCA sugli effetti del 5-FU era dose dipendente (Figura 1). L’interazione tra 5-FU e DCA è stata analizzata con il metodo degli effetti mediani. La combinazione di DCA e 5-FU ha prodotto effetti sinergici o additivi a seconda dell’intervallo del livello di uccisione cellulare (Fa). Tutte e quattro le linee cellulari di CRC hanno mostrato effetti sinergici con 5-FU e DCA. Il sinergismo è stato statisticamente significativo in LS174t a livelli di inibizione del 50% e dell’80%, raggiungendo valori di CI compresi tra 0,46 e 0,61, e nella linea cellulare HT-29 a livelli di inibizione del 50 e dell’80%, con valori di CI compresi tra 0,42 e 0,52. Nelle cellule SW620 e LoVo, i valori di IC ai livelli di inibizione del 50% e dell’80% erano rispettivamente 0,64-0,75 e 0,75-0,94 (Figura 2).
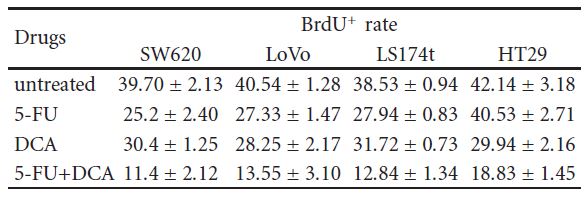
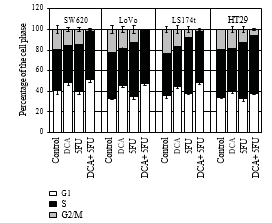
3.3. Il DCA ha aumentato l’efficacia dell’effetto antiproliferativo del 5-FU. Per confermare che la diminuzione della vitalità cellulare era dovuta a una ridotta proliferazione, sono state utilizzate l’immunocitochimica e la citometria a flusso. Le cellule sono state trattate con 10 mM di DCA in combinazione con 20 μM di 5-FU. Come previsto, le cellule trattate con DCA hanno dimostrato una ridotta proliferazione rispetto alle cellule non trattate. Il numero di cellule BrdU positive su quattro linee cellulari CRC dopo il trattamento con 5-FU e DCA è stato rispettivamente di 11,4 ± 2,12, 13,55 ± 3,10, 12,84 ± 1,34 e 18,83 ± 1,45, inferiore a quello trattato con 5-FU o DCA da soli (P < .01, vedi Tabella 2). Inoltre, il trattamento con DCA ha potenziato l’arresto del ciclo cellulare nella fase G1. Quando sono state trattate con DCA e 5-FU, le cellule bloccate nella faseG1/S erano più di quelle incorporate con DCA o 5-FU da soli (Figura 3).
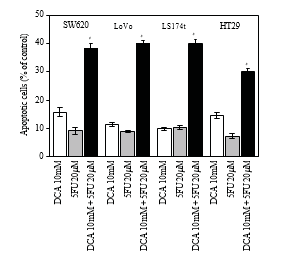
3.4. Aumento dell’apoptosi indotta da DCA combinato con 5-FU nelle cellule CRC. Per indagare ulteriormente sulla diminuzione della vitalità delle cellule CRC trattate con un regime di combinazione, l’apoptosi è stata determinata mediante citometria a flusso. Come mostrato nella Figura 4, il DCA da solo ha aumentato la percentuale di apoptosi delle cellule CRC. Quando sono state trattate con 10 mM di DCA, i tassi di apoptosi di quattro linee cellulari CRC sono stati rispettivamente del 15,72 ± 1 ,63%, 11,32 ± 0 ,74%, 9,77 ± 0 ,53% e 14,52 ± 1 ,00%, mentre i tassi di apoptosi Tabella 1: I valori IC50 (concentrazioni necessarie per ridurre la vitalità delle cellule del 50% rispetto alle cellule di controllo) sono stati calcolati utilizzando una regressione lineare o non lineare (funzione Hill a tre parametri) (R2> 0 ,9). Sono presentati come media ± SD di almeno tre esperimenti indipendenti. Valori IC50 dei farmaci studiati per l’inibizione della crescita di varie linee cellulari (le cellule sono state incubate con i farmaci per 48 ore).
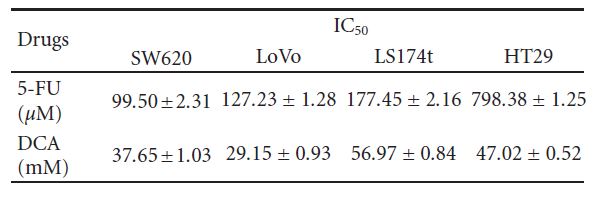
Tabella 2: le cellule SW620, LoVo, LS174t, HT29 e i controlli non cancerosi 293T sono stati trattati con 10 mM di DCA e 20 μM di 5-FU, da soli o in combinazione, per 48 ore, e poi sono stati pulsati con BrdU. Le cellule sono state quindi raccolte e colorate e il numero di cellule BrdU+ è stato calcolato come numero medio di cellule positive in otto campi visivi differenti in un’immagine (ingrandimento, 400X). Questo calcolo è stato ripetuto tre volte. ∗P< .05, rispetto al controllo. Cellule BrdU+ in trattamenti farmacologici differenti (%, media ± SD).
erano 9,14 ± 119%, 8,82 ± 0 ,41%, 10,31 ± 0 ,71% e 7,27±0 ,96% con il 5-FU. Quando è stata applicata la combinazione di DCA e 5-FU, il tasso di apoptosi è stato molto più alto rispetto al 5-FU o al DCA da soli (P < .05), il che indica che l’effetto apoptotico è stato aumentato tramite la combinazione di DCA e 5-FU (Figura 4).
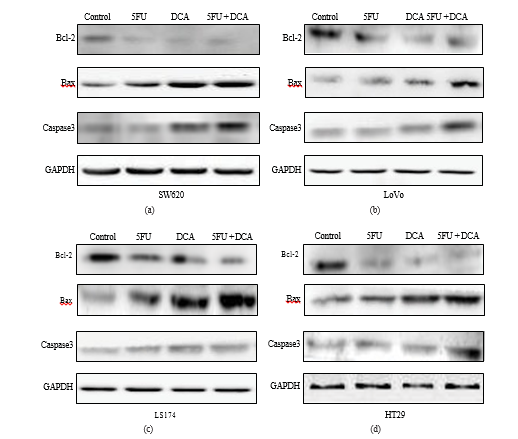
3.5. Cambiamenti nelle molecole associate all’apoptosi stimolate da DCA e 5-FU. Per confermare che l’aumento dell’apoptosi indotto dalla terapia combinata è dovuto alla modifica dell’espressione delle molecole associate all’apoptosi, è stato eseguito un saggio di western blot. Nella Figura 5, i risultati hanno indicato che il 5-FU o il DCA hanno ridotto l’espressione di Bcl-2 in quattro linee cellulari CRC rispetto ai controlli con PBS, e la combinazione di DCA e 5-FU ha ridotto significativamente l’espressione di Bcl-2 rispetto al DCA o al 5-FU da soli. Al contrario, le espressioni di Bax e caspasi-3 sono risultate significativamente aumentate nelle quattro linee cellulari CRC trattate con la combinazione di DCA e 5-FU rispetto al loro utilizzo singolo. L’aumento più evidente dell’espressione di Bax è stato rilevato nelle cellule LS174t, mentre nelle LoVo è risultato maggiore l’aumento dell’espressione della caspasi-3 (Figura 5).
4. Discussione
Nel presente studio, abbiamo dimostrato che il DCA non solo ha ridotto la vitalità e la proliferazione cellulare, ma ha anche un’efficienza antitumorale sinergica con l’agente chemioterapico 5-FU in vitro nelle cellule CRC.
Il DCA non ha invece effetti significativi sulle cellule non cancerose. Inoltre, abbiamo dimostrato che l’apoptosi indotta dal DCA contribuisce al suo effetto antitumorale sinergico. Rispetto al DCA o al 5-FU da soli, l’uso combinato di questi due farmaci promuove l’apoptosi delle cellule CRC. il 5-FU è un farmaco chemioterapico utilizzato per trattare diversi tipi di cancro, tra cui quello del colon-retto, della mammella, dell’esofago e dello stomaco [15]. Tuttavia, la tossicità legata al 5-FU è un problema serio e comune per molti pazienti oncologici, con la mielosoppressione e la tossicità gastrointestinale che sono gli effetti collaterali più comunemente osservati [16]. L’attività clinica del 5-FU è modesta alle dosi standard e, in generale, il dosaggio è limitato dal profilo di sicurezza. Di conseguenza, ci troviamo di solito di fronte al dilemma di decidere la dose terapeutica di 5-FU. Sono state sviluppate diverse strategie per migliorare l’attività clinica del 5-FU, come la modulazione biochimica [17], la modifica del calendario di somministrazione [18] e l’uso di una terapia combinata [19-22]. Il DCA è una piccola molecola inodore, incolore, poco costosa e relativamente non tossica. È in uso clinico dal 1969 per il trattamento dell’acidosi lattica attraverso il potenziamento della capacità dei mitocondri di generare energia e la riduzione dell’accumulo di acido lattico [23].
Il DCA è stato considerato un potenziale regime terapeutico antitumorale da quando un gruppo canadese ha scoperto che causava la regressione di diversi tumori, tra cui quelli al polmone, al seno e al cervello [24, 25]. Quando viene somministrato alle cellule tumorali, queste passano dalla glicolisi alla produzione di energia mitocondriale. Inoltre, i mitocondri funzionali aiutano le cellule a riconoscere le anomalie funzionali e a innescare la morte cellulare grazie all’inibizione della crescita tumorale e all’induzione dell’apoptosi in alcuni tipi di cancro. Nel presente studio abbiamo riscontrato lo stesso effetto di inibizione della vitalità nei CRC, che è risultato dose-dipendente e diverso a seconda del grado di differenziazione del tumore.
I risultati hanno anche indicato che il DCA a basso dosaggio esercita un effetto sinergico con l’agente chemioterapico 5-FU nell’arrestare la crescita delle cellule CRC, analizzata quantitativamente secondo il metodo Chou-Talalay.
I risultati della proliferazione cellulare hanno inoltre dimostrato che il DCA ha potenziato gli effetti antiproliferativi del 5-FU. Il numero di cellule positive al BrdU è diminuito quando sono state trattate con 5-FU o DCA, mentre le cellule macchiate di BrdU sono diminuite significativamente quando sono state trattate con la combinazione di 5-FU e DCA rispetto al loro uso singolo. L’inibizione della crescita delle cellule CRC è stata accompagnata da un arresto del ciclo cellulare. La combinazione di DCA e 5-FU ha indotto l’arresto del ciclo cellulare in faseG1/S nelle linee cellulari CRC, mentre l’arresto in faseG1 indotto dal 5-FU non era evidente. L’induzione dell’arresto del ciclo cellulare può derivare dall’inibizione della capacità di sintetizzare o riparare il DNA, che può portare all’apoptosi cellulare.
Il DCA sembra esercitare effetti biochimici coerenti con l’inversione dell’effetto di Warburg e l’uccisione delle cellule tumorali. Abbiamo scoperto che il DCA ha indotto l’apoptosi delle cellule CRC, in linea con gli studi precedenti sul DCA. È importante notare che la combinazione del DCA con il 5-FU ha aumentato il numero di cellule apoptotiche rispetto al solo 5-FU, dimostrando che il DCA inibisce la crescita cellulare attraverso l’apoptosi.
Molti fattori che mediano l’apoptosi convergono nell’attivare l’effettore critico, la caspasi-3, che è considerata la proteasi chiave della famiglia delle caspasi nell’apoptosi delle cellule di mammifero [26]. Esiste sempre come precursore inattivo di 23 kD nel citoplasma, che viene attivato durante l’apoptosi e partecipa all’apoptosi indotta da molteplici fattori. La via dell’apoptosi dipendente dalla caspasi comprende principalmente la via dei mitocondri, dei recettori di morte e del reticolo endoplasmatico [27].
Il percorso mitocondriale è controllato e regolato dalla famiglia di proteine Bcl-2 [28, 29], che si dividono in due parti, i membri antiapoptotici (Bcl-2) e proapoptotici (Bax) [30]. Studi recenti indicano che Bcl-2 inibisce l’apoptosi attraverso l’inibizione della rimozione di Bax alla membrana esterna mitocondriale [31]. Abbiamo studiato l’espressione di caspasi-3, Bcl-2 e Bax a livello proteico e i risultati del western blot hanno mostrato che le espressioni di caspasi-3 e Bax erano aumentate, mentre l’espressione di Bcl-2 era diminuita nel trattamento combinato di DCA e 5-FU, rispetto al trattamento singolo. Questi risultati hanno suggerito che l’apoptosi indotta dalla combinazione di DCA e 5-FU potrebbe essere correlata alla via mitocondriale caspasi-dipendente. Precedenti indagini hanno suggerito che l’induzione dell’apoptosi da parte del DCA derivi dal ritorno della disfunzione dei mitocondri e della via NFAT-Kv1.5 [9, 12], che si concentravano sullo stesso punto del presente studio, ovvero l’apoptosi mediata dai mitocondri.
Riconoscimento
Questo lavoro è stato sostenuto dalla National Natural Science Foundation of China (NSFC, n. 30873015). J. Tong e G. Xie hanno contribuito in egual misura a questo lavoro.
Riferimenti
[1] A. Jemal, R. Siegel, E. Ward, Y. Hao, J. Xu e M. J. Thun, “Cancer statistics, 2009”, CA Cancer Journal for Clinicians, vol. 59, no. 4, pp. 225-249, 2009.[2] J. A. Meyerhardt e R. J. Mayer, “Drug therapy: systemic therapy for colorectal cancer”, The New England Journal of Medicine, vol. 352, no. 5, pp. 476-487, 2005.
[3] N. C. Tebbutt, E. Cattell, R. Midgley, D. Cunningham e D. Kerr, “Systemic treatment of colorectal cancer”, European Journal of Cancer, vol. 38, n. 7, pp. 1000-1015, 2002.
[4] M. Gusella, A. C. Frigo, C. Bolzonella et al., “Predittori di sopravvivenza e tossicità in pazienti in terapia adiuvante con 5-fluorouracile per il cancro del colon-retto”, British Journal of Cancer, vol. 100, n. 10, pp. 1549-1557, 2009.
[5] D. Hanahan e R. A. Weinberg, “The hallmarks of cancer”, Cell, vol. 100, n. 1, pp. 57-70, 2000.
[6] Z. Chen, W. Lu, C. Garcia-Prieto e P. Huang, “TheWarburg effect and its cancer therapeutic implications”, Journal of Bioenergetics and Biomembranes, vol. 39, no. 3, pp. 267-274, 2007.
[7] Y. Chen, R. Cairns, I. Papandreou, A. Koong, andN. C. Denko, “Oxygen consumption can regulate the growth of tumors, a new perspective on theWarburg effect”, PLoS ONE, vol. 4, n. 9, Article ID e7033, 2009.
[8] A. Aynsley Green, A. M. Weindling, G. Soltesz e P. A. Jenkins, “Acidosi lattica transitoria e iperalaninemia associate all’ipoglicemia iperinsulinemica neonatale: gli effetti del dicloroacetato (DCA)”, European Journal of Pediatrics, vol. 141, n. 2, pp. 114-117, 1983.
[9] J. Y. Y. Wong,G. S. Huggins, M. Debidda, N. C. Munshi e I. De Vivo, “Il dicloroacetato induce l’apoptosi nelle cellule del cancro endometriale”, Oncologia ginecologica, vol. 109, no. 3, pp. 394-402, 2008.
[10] W. Cao, S. Yacoub, K. T. Shiverick et al., “Il dicloroacetato (DCA) sensibilizza alle radiazioni sia le cellule di cancro alla prostata wild-type che quelle che esprimono il bcl-2 in vitro”, Prostate, vol. 68, n. 11, pp. 1223-1231, 2008.
[11] E. D. Michelakis, L. Webster e J. R. Mackey, “Il dicloroacetato (DCA) come potenziale terapia a bersaglio metabolico per il cancro”, British Journal of Cancer, vol. 99, n. 7, pp. 989-994, 2008.
[12] S. Bonnet, S. L. Archer, J. Allalunis-Turner et al., “A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth”, Cancer Cell, vol. 11, n. 1, pp. 37-51, 2007.
[13] T. C. Chou e P. Talalay, “Analysis of combined drug effects: a new look at a very old problem”, Trends in Pharmacological Sciences, vol. 4, pp. 450-454, 1983.
[14] T. C. Chou e P. Talalay, “Analisi quantitativa delle relazioni dose-effetto: gli effetti combinati di più farmaci o inibitori enzimatici”, Advances in Enzyme Regulation, vol. 22, pp. 27-55, 1984.
[15] D. B. Longley, D. P. Harkin e P. G. Johnston, “5-Fluorouracile: meccanismi d’azione e strategie cliniche”, Nature Reviews Cancer, vol. 3, no. 5, pp. 330-338, 2003.
[16] M. W. Saif, A. Choma, S. J. Salamone e E. Chu, “Pharmacokinetically guided dose adjustment of 5-fluorouracil: a rational approach to improving therapeutic outcomes”, Journal of the National Cancer Institute, vol. 101, n. 22, pp. 1543-1552, 2009.
[17] C.G. Leichman, K. Chansky, J. S. Macdonaldet al., “Biochemical modulation of 5-fluorouacil through dihydropyrimidine dehydrogenase inhibition: a Southwest OncologyGroup phase II trial of eniluracil and 5-fluorouracil in advanced resistant colorectal cancer,” Investigational New Drugs, vol. 20, no. 4, pp. 419-424, 2002.
[18] F. A. Levi, R. Zidani, J. M. Vannetzel et al., “Chronomodulated versus fixed-infusion-rate delivery of ambulatory chemotherapy with oxaliplatin, fluorouracil, and folinic acid (leucovorin) in patients with colorectal cancer metastases: a randomized multi-institutional trial”, Journal of the National Cancer Institute, vol. 86, no. 21, pp. 1608-1617, 1994.
[G. Melen-Mucha, E. Balcerczak, S. Mucha, M. Panczyk, S. Lipa e M. Mirowski, “Expression of p65 gene in experimental colon cancer under the influence of 5-fluorouracil given alone and in combination with hormonal modulation”, Neoplasma, vol. 51, n. 4, pp. 319-324. 4, pp. 319-324, 2004.
[20] F. Richards II, L. D. Case e D. R. White, “Chemioterapia combinata (5-fluorouracile, metil-CCNU, mitomicina C) rispetto al solo 5-fluorouracile per il carcinoma colorettale avanzato non trattato in precedenza. Uno studio di fase III della piedmont oncology association”, Journal of Clinical Oncology, vol. 4, no. 4, pp. 565-570, 1986.
[21] A. Aquino, S. P. Prete, J. W. Greiner et al., “Effetto del trattamento combinato con 5-fluorouracile, γ-interferone o acido folinico sull’espressione dell’antigene carcinoembrionale nelle cellule del cancro del colon”, Clinical Cancer Research, vol. 4, n. 10, pp. 2473-2481, 1998.
[22] S. Obi, H. Yoshida, R. Toune et al., “Terapia combinata di 5-fluorouracile intraarterioso e interferone-alfa sistemico per il carcinoma epatocellulare avanzato con invasione venosa portale”, Cancer, vol. 106, n. 9, pp. 1990-1997, 2006.
[23] P. W. Stacpoole, “Review of the pharmacologic and therapeutic effects of diisopropylammonium dichloroacetate (DIPA)”, The Journal of Clinical Pharmacology, vol. 9, no. 5, pp. 282-291, 1969.
[24] S. Bonnet, S. L. Archer, J. Allalunis-Turner et al., “A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth”, Cancer Cell, vol. 11, n. 1, pp. 37-51, 2007.
[25] E. D.Michelakis, G. Sutendra, P.Dromparis et al., “Metabolic modulation of glioblastoma with dichloroacetate”, Science Translational Medicine, vol. 2, no. 31, pp. 31-ra34, 2010.
[26] T. Fernandes-Alnemri, G. Litwack e E. S. Alnemri, “CPP32, una nuova proteina apoptotica umana con omologia alla proteina di morte cellulare Ced-3 di Caenorhabditis elegans e all’enzima di conversione dell’interleuchina-1β dei mammiferi”, The Journal of Biological Chemistry, vol. 269, no. 49, pp. 30761-30764, 1994.
[27] H. Mehmet, “Le caspasi trovano un nuovo posto dove nascondersi”, Nature, vol. 403, n. 6765, pp. 29-30, 2000.
[28] E. Yang e S. J. Korsmeyer, “Tanatopsia molecolare: un discorso sulla famiglia BCL2 e la morte cellulare”, Blood, vol. 88, n. 2, pp. 386-401, 1996.
[29] D. R. Green e J. C. Reed, “Mitocondri e apoptosi”, Science, vol. 281, no. 5381, pp. 1309-1312, 1998.
[30] J. C. Reed, “Doppia identità per le proteine della famiglia Bcl-2”, Nature, vol. 387, n. 6635, pp. 773-776, 1997.
[31] B. Antonsson, S. Montessuit, B. Sanchez e J. C. Martinou, “Bax è presente come oligomero/complesso ad alto peso molecolare nella membrana mitocondriale delle cellule apoptotiche”, The Journal of Biological Chemistry, vol. 276, n. 15, pp. 11615- 11623, 2001.