Cecilie Abildgaard1, Christina Dahl1, Ahmad Abdul-Al1, Annette Christensen1 et Per Guldberg1
1 Centre de recherche de la Société danoise du cancer, Copenhague, Danemark
Correspondance : Per Guldberg, courriel : [email protected]
Reçue : 19 avril 2017
Accepté : 19 juillet 2017
Publié : 24 août 2017
Résumé
La dérégulation du métabolisme pendant la progression du mélanome est étroitement associée à l’acquisition d’altérations génétiques et épigénétiques dans les régulateurs des voies métaboliques. Le récepteur de l’acide rétinoïque bêta (RARβ) est épigénétiquement réduit au silence dans une grande proportion de mélanomes, mais un lien entre RARβ et le recâblage métabolique du mélanome n’a pas été établi. Ici, nous montrons que dans les mélanocytes humains primaires, l’acide rétinoïque all-trans (un agoniste de RARβ) induit une inhibition de la croissance accompagnée d’une diminution du métabolisme glycolytique et oxydatif, tandis que l’inhibition sélective de RARβ conduit à une augmentation du taux glycolytique basal et à une sensibilité accrue à l’inhibition de la glycolyse. Dans les cellules de mélanome, l’inhibition de RARβ a favorisé une baisse de la respiration mitochondriale et une augmentation de l’activité glycolytique, ce qui a entraîné un stress énergétique et l’activation du capteur d’énergie qu’est la protéine kinase activée par l’AMP. Ce changement métabolique a augmenté la sensibilité à l’inhibition glycolytique et à la stimulation du métabolisme mitochondrial par le dichloroacétate, un inhibiteur de la pyruvate déshydrogénase kinase. Dans les cellules de mélanome hébergeant la mutation BRAFV600E, l’activation de RARβ a antagonisé l’effet de l’inhibiteur de BRAF PLX4032 (vemurafenib). Collectivement, ces données suggèrent que la signalisation RARβ est impliquée dans la régulation du métabolisme cellulaire dans le mélanome et pourrait constituer une cible potentielle dans les stratégies de traitement combiné.
Mots clés : mélanome, métabolisme du cancer, récepteur de l’acide rétinoïque β, respiration mitochondriale, dichloroacétate
Abréviations : ATRA : acide rétinoïque all-trans ; DCA : dichloroacétate ; ECAR : taux d’acidification extracellulaire ; OCR : taux de consommation d’oxygène ; ROS : espèces réactives de l’oxygène
abildgaard et al. Il s’agit d’un article en accès libre distribué selon les termes de la licence Creative Commons Attribution License 3.0 (CC BY 3.0), qui permet l’utilisation, la distribution et la reproduction sans restriction sur n’importe quel support, à condition que l’auteur original et la source soient mentionnés.
INTRODUCTION
Le mélanome, la forme la plus mortelle de cancer de la peau, est responsable de 50 000 décès par an et son incidence continue d’augmenter dans le monde entier. Alors que le mélanome cutané primaire est curable par chirurgie, la forme la plus avancée de la maladie (stade IV) est associée à une survie à 10 ans de 10-15% [1], reflétant sa résistance notoire aux thérapies anticancéreuses conventionnelles. Les récentes avancées thérapeutiques incluent les inhibiteurs de points de contrôle immunitaire et les thérapies ciblant les oncogènes ou les effecteurs en aval de la voie MAPK (par exemple, les inhibiteurs de BRAF et MEK). Cependant, le développement d’une résistance acquise aux médicaments conduit finalement à une rechute dans la majorité des cas [2, 3].
Le mélanome se développe à partir de cellules productrices de mélanine, appelées mélanocytes, par l’acquisition de multiples altérations génomiques. Les facteurs les plus courants du mélanome comprennent des mutations activatrices de BRAF et NRAS et des mutations inactivatrices ou des délétions de CDKN2A (codant pour p16INK4A et p14ARF), PTEN et TP53 [4]. Des preuves récentes suggèrent qu’une fonction commune partagée par certains de ces gènes est le contrôle du métabolisme cellulaire [5, 6]. Au cours de la progression du mélanome, le métabolisme cellulaire est reprogrammé, ce qui implique un passage de la respiration mitochondriale à la glycolyse aérobie, entraînant une augmentation de la consommation de glucose et de la production d’acide lactique (effet Warburg) [7]. Plusieurs rapports basés sur des modèles in vitro et in vivo de mélanome et des études cliniques de patients atteints de mélanome ont démontré un lien entre les mutations activatrices au codon V600 de BRAF (le plus souvent BRAFV600E) et la glycolyse aérobie [8-10]. Au niveau moléculaire, BRAFV600E régule la phosphorylation oxydative en supprimant le maître régulateur de la biogénèse mitochondriale, PGC1α, par l’inhibition du facteur de transcription associé à la microphtalmie (MITF). En revanche, l’inhibition de BRAFV600E entraîne une dépendance oxydative par l’induction de PGC1α et l’augmentation de la respiration mitochondriale [11]. La diminution correspondante de l’activité glycolytique peut être visualisée par TEP-CT chez les patients atteints de mélanome traités par des inhibiteurs de BRAF, montrant une absorption réduite du glucose dans le tissu tumoral [10]. Les essais cliniques de phase III de l’inhibiteur BRAFV600E vemurafenib (PLX4032) ont montré une amélioration de la survie globale et sans progression chez les patients atteints de mélanome métastatique [12]. Les inhibiteurs mitochondriaux ont été suggérés comme des adjuvants utiles aux inhibiteurs de la voie BRAF pour améliorer l’effet ou prévenir le développement de la résistance aux médicaments [13-15].
Outre les facteurs génétiques bien caractérisés, le génome du mélanome contient de nombreuses altérations épigénétiques. L’une des cibles épigénétiques récurrentes dans le mélanome est RARB codant pour le récepteur de l’acide rétinoïque bêta (RARβ), qui est réduit au silence par hyperméthylation du promoteur dans 45 à 70 % des mélanomes cutanés [16, 17]. Dans les cellules de la lignée mélanocytaire, RARβ est le médiateur de l’inhibition de la croissance induite par l’acide rétinoïque (vitamine A) et de la mélanogenèse, marqueur de la différenciation mélanocytaire [18]. Nous avons précédemment montré que l’activation de RARβ dans les mélanocytes induit une régulation à la hausse de p14ARF [17], qui protège contre le dysfonctionnement mitochondrial et le stress oxydatif [19]. Nous montrons ici que les mélanocytes humains répondent à l’activation de RARβ en réduisant le métabolisme oxydatif, potentiellement dans le cadre d’une réponse de différenciation. Dans les cellules de mélanome, l’activation de RARβ antagonise l’effet du PLX4032, tandis que l’inhibition de RARβ induit une dépendance glycolytique et un stress énergétique, rendant les cellules vulnérables au traitement par l’inhibiteur de la pyruvate déshydrogénase kinase, le dichloroacétate (DCA).
RÉSULTATS
L’activation de RARβ réduit la croissance et le taux métabolique des mélanocytes
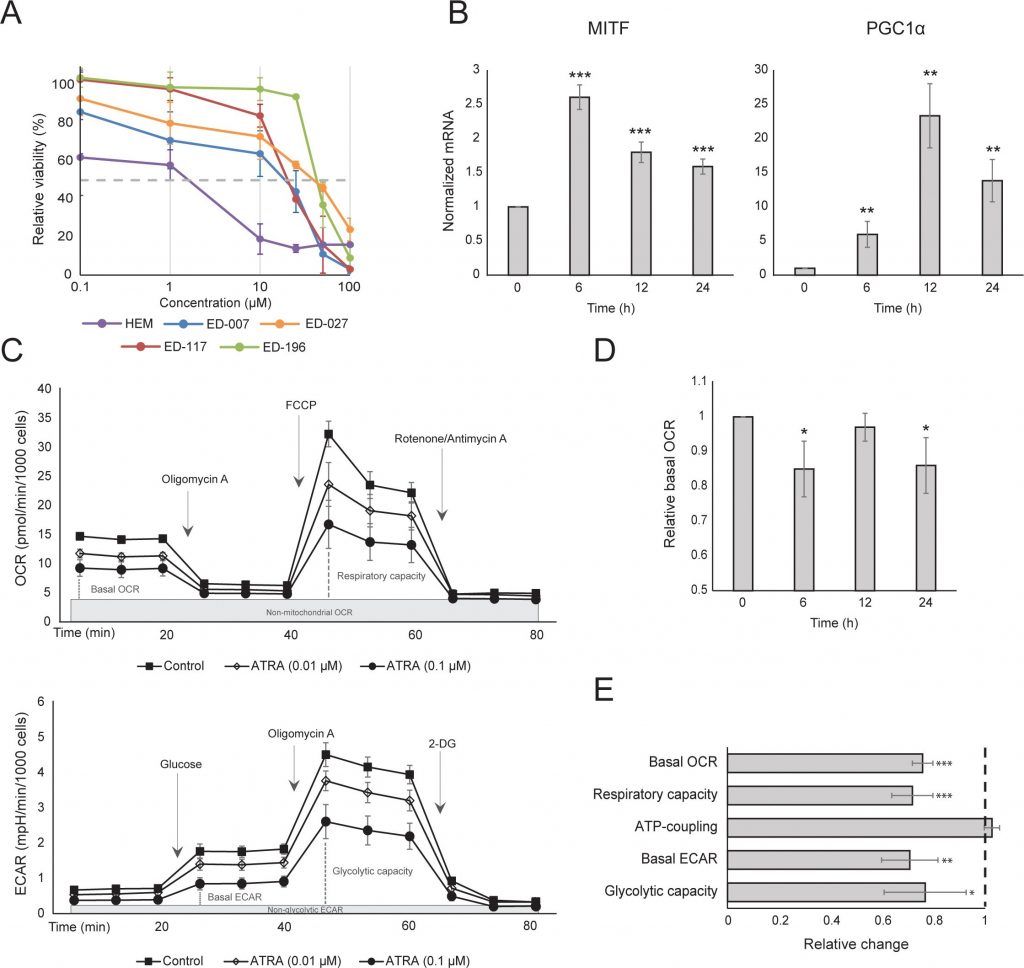
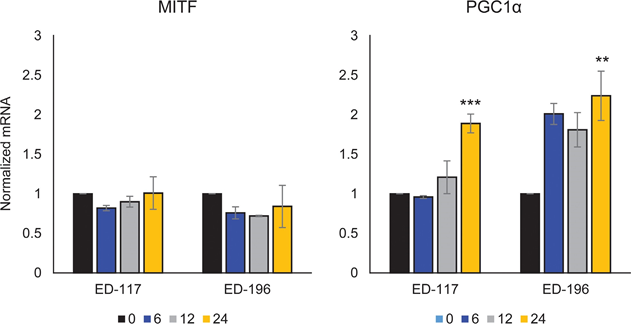
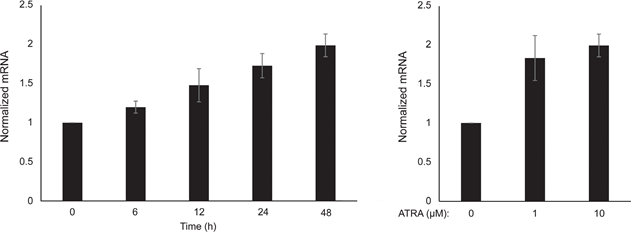
Nous avons d’abord déterminé l’effet de l’activation de RARβ sur la croissance des mélanocytes épidermiques humains primaires. Les cellules ont été traitées avec l’agoniste RARβ, l’acide rétinoïque all-trans (ATRA), pendant 6 jours, et la réponse de croissance a été déterminée avec un test de viabilité à base de cristal violet. Conformément aux rapports précédents [17, 20, 21], l’ATRA a réduit la croissance des mélanocytes de manière dose-dépendante (figure 1A), avec une CI50 de 2,4 μM (tableau 1). Il a déjà été montré que le traitement à court terme (<24 h) avec l’atra induit la différenciation et la mélanogenèse dans les mélanocytes, tandis que l’exposition à long terme (>24 h) réduit la prolifération et induit l’apoptose [20, 21]. Nous avons constaté que l’ATRA (0,1 μM) induisait une régulation transitoire à la hausse du facteur de transcription MITF (microphthalmia-associated transcription factor) spécifique à la lignée mélanocytaire, avec un pic d’expression après 6 h, puis un déclin vers les niveaux basaux (figure 1B). Dans les cellules de mélanome, MITF régule l’expression de PGC1α, un marqueur d’un phénotype oxydatif [22]. Nous avons donc étudié l’expression de PGC1α dans les mélanocytes à différents moments après exposition à l’ATRA (0,1 μM). Comme le montre la figure 1B, PGC1α a également été régulé transitoirement à la hausse, avec un retard de ~6 heures par rapport à MITF.
| Cellules/lignées cellulaires | Cellules/lignes cellulaires | Caractéristiques | Caractéristiques | Caractéristiques | Valeurs IC50 | Valeurs IC50 | Valeurs de la CI50 | Valeurs de la CI50 |
|---|---|---|---|---|---|---|---|---|
| Numéro ED | Nom | Statut BRAF* | Expression de RARβ ** | expression de p14ARF | ATRA (μM) | LE135 (μM) | DCA (mM)*** | PLX4032 (μM) |
| HEM# | WT | + | + | 2.4±1.6 | 2.8±0.8 | 69.1±6.4 | NA | |
| ED-007 | FM-3 | WT | + | – | 18.6±8.7 | 8.6±1.0 | 12.2±2.2 | NA |
| ED-027 | FM-82 | BRAFV600E | + | + | 39.8±5.3 | 10.7±1.3 | 17.7±2.1 | 0.52±0.04 |
| ED-117 | Mel-NT3-00 | BRAFV600E | + | + | 25.5±5.0 | NA | 37.6 ±2.2 | 0.51±0.09 |
| ED-196 | Ma-Mel-51 | BRAFV600E | + | + | 46.2±9.1 | 8.4±0.4 | 35.8±3.2 | 0.26±0.06 |
Les valeurs IC50 représentent la moyenne ± l’écart-type de ≥3 expériences indépendantes.
*Confirmépar pyroséquençage
**Confirmépar qPCR
***ValeursIC50 publiées par Abildgaard et al. [29]
#Mélanocytes épidermiques humains
En raison du rôle de PGC1α dans la biogenèse mitochondriale, nous avons ensuite cherché à savoir si l’expression de PGC1α était corrélée au niveau de la respiration mitochondriale. En utilisant l’instrument Seahorse XFe96, nous avons mesuré le taux de consommation d’oxygène (OCR) et le taux d’acidification extracellulaire (ECAR), qui sont des indicateurs du taux de respiration mitochondriale et de l’activité glycolytique, respectivement. L’OCR et l’ECAR ont été mesurés pendant l’ajout séquentiel de modulateurs métaboliques, ce qui a permis de déterminer les taux et les capacités basales des deux systèmes énergétiques (Figure 1C). Pour mieux comprendre la dépendance temporelle des réponses à l’ATRA, les paramètres métaboliques ont été mesurés après des expositions à court terme (6-24 h) et à long terme (7 jours). Lors du traitement des mélanocytes avec l’ATRA (0,1 μM) pendant 6 ou 24 h, le ROC basal a été réduit. Cependant, après 12 h de traitement, le ROC était similaire aux niveaux de base (figure 1D). Ces fluctuations de l’état métabolique coïncidaient avec des modifications de l’expression de MITF et de PGC1α. L’exposition à long terme (7 jours) à une faible dose d’ATRA (0,01 μM) a entraîné une nouvelle diminution de l’OCR basal et de la capacité respiratoire (figure 1E).
Le couplage ATP mitochondrial n’a pas été affecté par l’ATRA (figure 1E), ce qui a entraîné une diminution nette de la production d’ATP par les mitochondries. Il a été démontré que PGC1α régulait à la hausse la protéine de découplage 2 (UCP2), conduisant à un léger découplage mitochondrial [23, 24]. L’expression d’UCP2 dans les mélanocytes n’a pas été affectée par le traitement par l’ATRA (figure supplémentaire 1), ce qui confirme que le couplage ATP n’a pas été modifié pendant l’activation de RARβ. L’expression des marqueurs de l’activité mitochondriale (COX5A, ATP5g1 et NDUFS3) et le contenu en ADN mitochondrial n’ont pas non plus été affectés par le traitement à l’ATRA (0,1 μM) jusqu’à 24 et 48 h, respectivement (figures supplémentaires 1 et 2).
n’a montré aucune signification statistique (NS).
Il n’y a pas eu de changement dans le taux glycolytique après 24 h de traitement par l’ATRA (données non présentées) ; toutefois, après 7 jours, l’activité glycolytique basale et la capacité glycolytique ont été considérablement réduites (figure 1E). La suppression des deux principaux systèmes énergétiques cellulaires indique que les mélanocytes présentent une demande énergétique plus faible en présence de l’ATRA, ce qui pourrait être la conséquence d’une croissance cellulaire réduite.
L’inhibition de RARβ augmente le taux glycolytique basal et favorise la dépendance glycolytique dans les mélanocytes
Undéfi lors de l’étude des effets cellulaires de l’ATRA est la présence de concentrations inconnues de vitamine A dans le sérum bovin fœtal, une source essentielle de micronutriments dans la plupart des milieux de culture cellulaire [25]. Pour étudier plus en détail le rôle de la signalisation RARβ dans le métabolisme des mélanocytes, nous avons donc utilisé l’antagoniste de RARβ LE135, qui cible RARβ avec une sélectivité modérée par rapport à RARα et une sélectivité élevée par rapport à RARγ et RXRα [26].
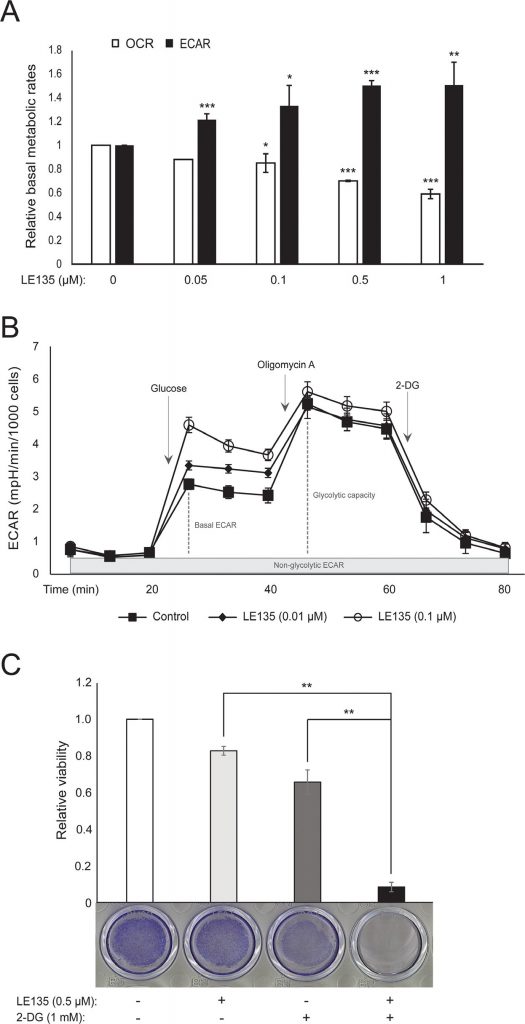
Nous avons répété les protocoles de l’hippocampe présentés dans la Figure 1C sur des mélanocytes traités avec différentes concentrations de LE135 pendant 24 h et 7 jours. Après 24 h, nous avons observé une augmentation dose-dépendante de l’activité glycolytique, avec une augmentation de l’ECAR basal jusqu’à 50%, et une réduction correspondante de l’OCR (Figure 2A). L’ECAR basale était toujours augmentée après 7 jours de traitement (Figure 2B). Il n’y a pas eu d’augmentation significative de la capacité glycolytique, ce qui suggère que les cellules ont été forcées de compter sur la glycolyse pour la production d’énergie. Cela a été confirmé par une plus grande sensibilité de ces cellules à l’inhibiteur de la glycolyse, le 2-désoxy-D-glucose (2-DG), en présence de LE135 (Figure 2C).
L’ATRA antagonise l’effet de l’inhibition de BRAF dans les cellules de mélanome
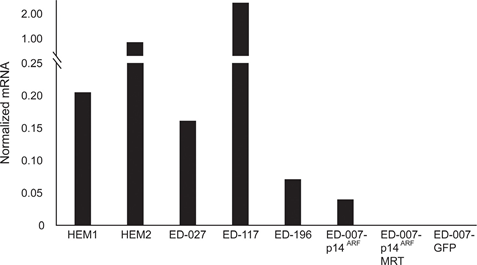
RARB est réduit au silence par hyperméthylation du promoteur dans de nombreux mélanomes, ce qui suggère qu’il possède des propriétés de suppression des tumeurs [16]. Dans une étude précédente des événements génétiques et épigénétiques dans 110 lignées de cellules de mélanome, nous avons trouvé une prévalence de 66% pour les mutations de BRAF et 45% pour l’hyperméthylation du promoteur de RARB, sans corrélation entre ces deux événements [17]. Pour étendre ces données, nous avons examiné l’expression de RARβ dans 84 de ces lignées cellulaires de mélanome ainsi que dans les mélanocytes épidermiques humains. Les niveaux d’expression variaient fortement entre les lignées cellulaires de mélanome, allant d’une absence totale d’expression à des niveaux jusqu’à 27 fois supérieurs à ceux des mélanocytes (Figure 3A). Comme prévu, l’hyperméthylation du promoteur de RARB était associée à des niveaux d’expression de RARβ faibles à indétectables, à quelques exceptions près. Il n’y avait aucune association entre les mutations BRAFV600E et les niveaux d’expression de RARβ (figure 3A), ce qui suggère que la sensibilité des cellules de mélanome à l’ATRA peut être indépendante du statut BRAF.
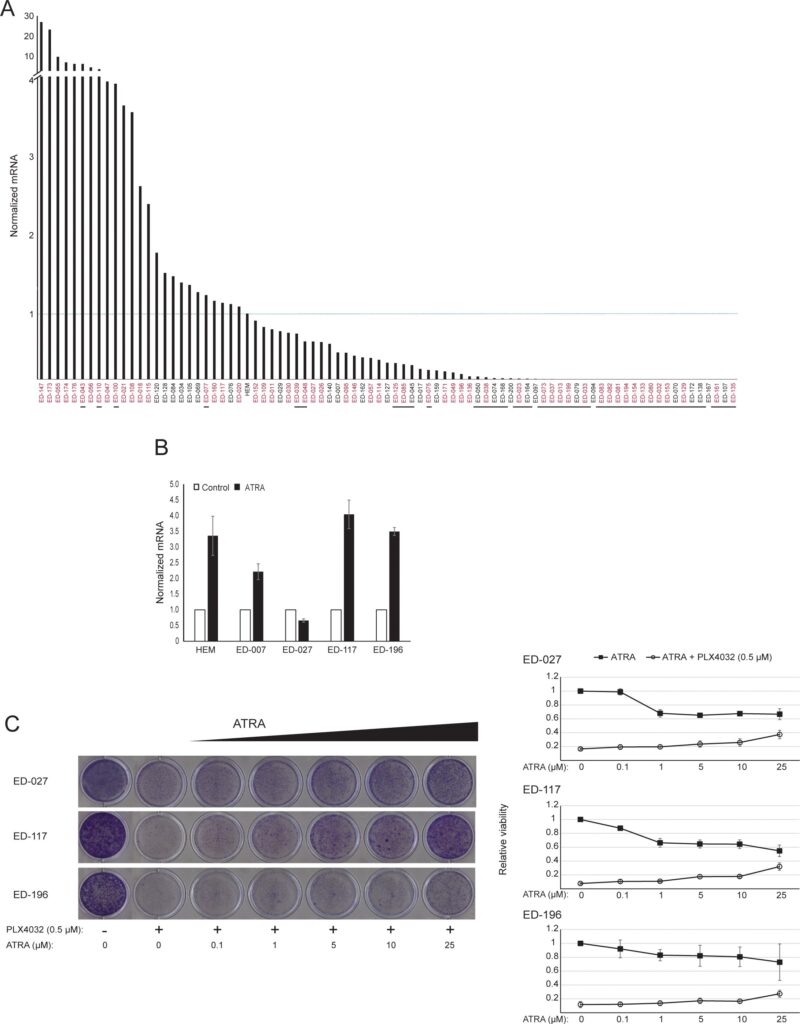
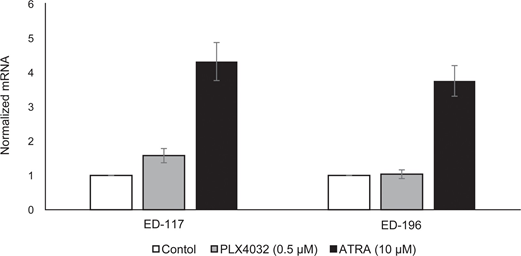
Pour étudier plus avant le rôle de la signalisation RARβ dans le mélanome, nous avons sélectionné quatre lignées cellulaires de mélanome RARβ-positives (ED-007, ED-027, ED-117 et ED-196) pour une analyse fonctionnelle. Trois de ces lignées cellulaires étaient mutées BRAFV600E (ED-027, ED-117 et ED-196) et une était de type sauvage BRAF (ED-007). Le traitement par ATRA a entraîné une diminution de la croissance des quatre lignées cellulaires (Figure 1A), bien que leur sensibilité soit inférieure à celle des mélanocytes, comme l’indiquent les valeurs de la CI50 (Tableau 1). L’expression de RARβ est connue pour être induite en réponse à l’ATRA [27]. Comme le montre la figure 3B, RARβ a été induit dans 3 des quatre lignées cellulaires de mélanome. Malgré une induction de RARβ plus prononcée dans ED-117 et ED-196, l’effet de l’ATRA sur l’expression de MITF et PGC1α a été atténué par rapport aux mélanocytes (figure supplémentaire 3). Il a été démontré précédemment que la suppression de l’axe MITF/PGC1α était une conséquence de l’activité oncogène de BRAF [11], ce qui pourrait contribuer à une réponse plus faible à l’ATRA. Conformément à cette notion, les cellules de type sauvage BRAF ont présenté la plus grande sensibilité à l’ATRA, bien qu’elle soit encore considérablement inférieure à celle des mélanocytes (tableau 1).
Afin d’étudier si le ciblage de BRAF avec PLX4032 restaurerait la sensibilité à l’ATRA, nous avons traité les lignées cellulaires de mélanome mutant BRAFV600E avec PLX4032 à une concentration proche des valeurs IC50 (cf. Tableau 1) en combinaison avec des concentrations croissantes d’ATRA (0,1-25 μM). De manière intéressante, l’ATRA a sauvé l’effet cytotoxique du PLX4032 dans toutes les lignées cellulaires. L’effet dose-dépendant de l’ATRA sur la croissance des cellules de mélanome traitées par le PLX4032 (figure 3C) indique un antagonisme entre les deux composés. Le traitement par PLX4032 (0,5 μM) n’a pas réduit l’expression de RARβ (figure supplémentaire 4), ce qui suggère un mécanisme différent pour cet antagonisme.
L’effet de l’ATRA sur le métabolisme cellulaire est affecté par le statut de p14ARF
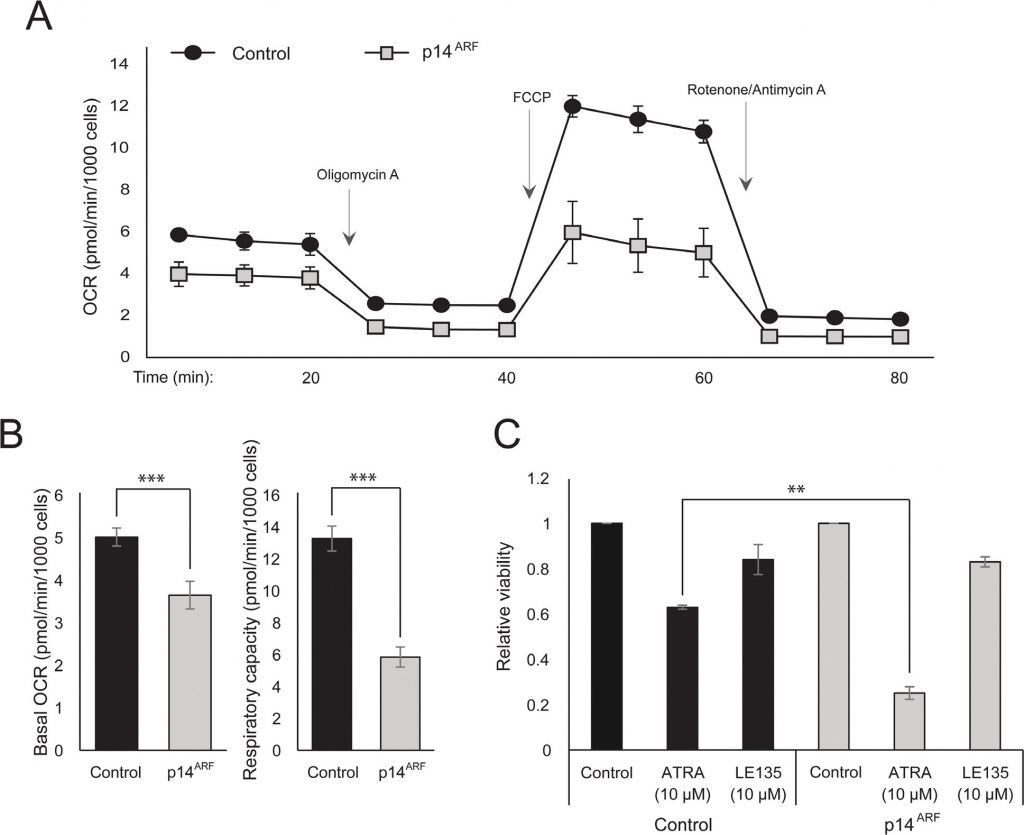
L’observation selon laquelle l’ATRA et la PLX4032 affectent tous deux la biogenèse mitochondriale pourrait indiquer une explication métabolique de l’effet antagoniste de ces composés. Il a été démontré précédemment que p14ARF est exprimé en tant que protéine cytoplasmique dans les mélanocytes normaux et qu’il protège ces cellules contre les mitochondries dysfonctionnelles [19]. Conformément aux résultats précédents [17], l’ATRA a augmenté l’expression de p14ARF dans les cellules de mélanome RARβ-positives (figure supplémentaire 5). Paradoxalement, bien que p14ARF soit fréquemment perdu dans les mélanomes par délétion du locus CDKN2A, il n’est pas possible d’éliminer de façon stable ARF dans les cellules de mélanome exprimant ce gène [17]; et données non présentées). Pour étudier le rôle potentiel de p14ARF dans la médiation d’une réponse cellulaire à l’ATRA, nous avons transfecté de façon stable la lignée cellulaire de mélanome ED-007 déficiente en p14ARF avec une construction EGFP-p14ARF. L’expression de p14ARF dans les cellules transfectées a été vérifiée par qPCR (figure supplémentaire 6). L’analyse de l’hippocampe a montré des profils métaboliques différents (figure 4A) avec un RCO basal et une capacité respiratoire significativement plus faibles dans les cellules avec une expression restaurée de p14ARF par rapport aux cellules transfectées de contrôle (figure 4B). De manière intéressante, les cellules exprimant p14ARF ont également montré une sensibilité accrue à l’ATRA (Figure 4C).
Leblocage de RARβ induit une dépendance glycolytique et un stress énergétique dans les cellules de mélanome et les sensibilise au dichloroacétate
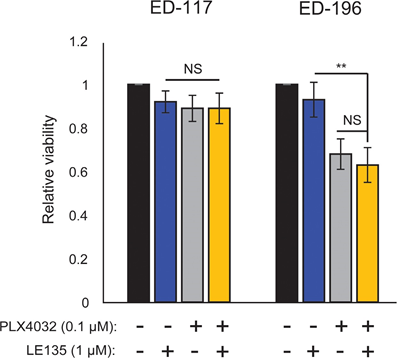
Sur la base de la découverte d’un effet antagoniste entre le PLX4032 et l’ATRA, nous avons testé l’effet combiné du PLX4032 et du LE135 pour une synergie potentielle dans les cellules de mélanome ED-117 et ED-196. Aucune inhibition coopérative de la croissance des mélanomes n’a été démontrée dans le montage expérimental utilisé ici (6 jours de traitement avec PLX4032 [0,1 μM] et LE135 [1 μM] ; figure supplémentaire 7).
Pour étudier plus en détail l’effet de l’inhibition de RARβ sur le métabolisme du mélanome, nous avons mesuré l’OCR et l’ECAR dans les lignées cellulaires de mélanome RARβ-positives traitées par LE135. De manière similaire à ce qui a été observé dans les mélanocytes (Figure 2A-2B), LE135 a augmenté le taux glycolytique basal et réduit la respiration mitochondriale basale dans les cellules de mélanome (Figure 5A-5B). Ce changement métabolique, qui est conforme à l’effet Warburg, était plus net dans les cellules de mélanome que dans les mélanocytes, ce qui a entraîné une augmentation du taux glycolytique de base pour atteindre la capacité maximale. Ceci a été illustré par l’incapacité d’augmenter davantage l’ECAR après l’ajout d’oligomycine A, indiquant un manque de flexibilité métabolique (Figure 5B). Pour étudier l’effet de LE135 sur la bioénergétique cellulaire, nous avons examiné l’état de phosphorylation de la protéine kinase activée par l’AMP (AMPK). L’AMPK détecte le statut énergétique cellulaire en réagissant à des niveaux élevés d’AMP, qui s’accumule lorsque le rapport ATP/ADP diminue. Ainsi, une réduction de la production d’énergie ou une augmentation de la consommation d’énergie peut favoriser une augmentation de l’AMP, qui se lie à l’AMPK et conduit à sa phosphorylation et à son activation [28]. Le traitement de lignées cellulaires de mélanome avec LE135 a entraîné la phosphorylation de l’AMPK, ce qui indique que les cellules subissent un stress énergétique. L’augmentation des niveaux de p-AMPK était déjà apparente après 2 heures et a continué à augmenter jusqu’à au moins 48 heures (Figure 5C). Ces résultats étaient différents de ceux obtenus dans les mélanocytes, où le traitement avec LE135 n’a pas entraîné l’activation de l’AMPK (figure supplémentaire 8). Les cellules de mélanome et les mélanocytes ont tous deux montré une réduction de la croissance cellulaire pendant 6 jours de traitement avec LE135 (valeurs IC50 indiquées dans le tableau 1). En outre, comme dans le cas des mélanocytes, LE135 a sensibilisé les cellules de mélanome à l’inhibition glycolytique par le 2-DG (Figure 5D).
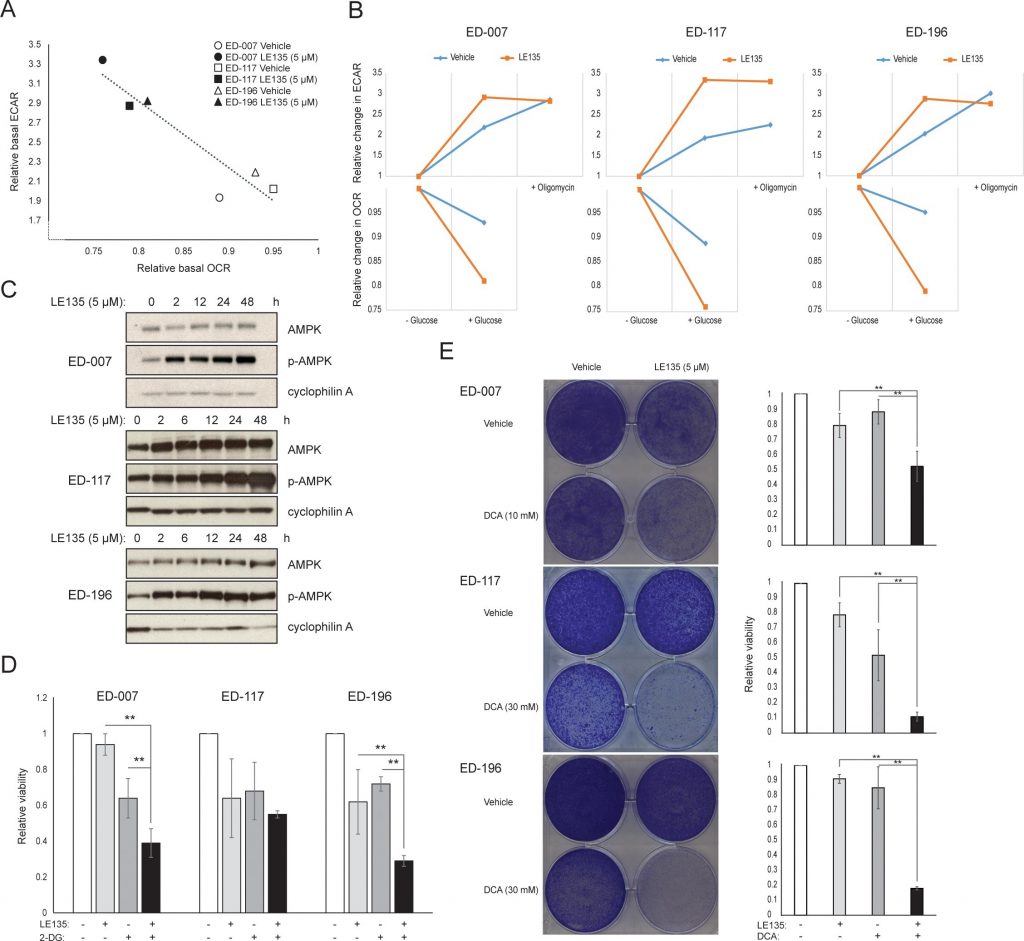
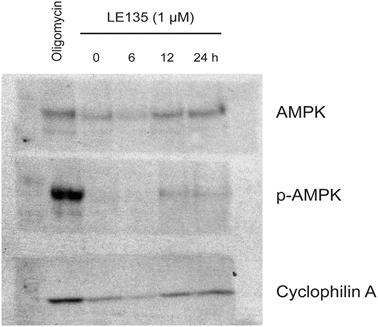
L’induction du stress énergétique avec LE135 dans les cellules de mélanome mais pas dans les mélanocytes fait allusion à une pertinence thérapeutique potentielle de ce composé dans les stratégies de traitement combiné. Il a déjà été démontré que l’inhibiteur de la pyruvate déshydrogénase kinase, le DCA, inhibe la croissance des cellules de mélanome en induisant une modification du métabolisme au détriment de la glycolyse, rendant les cellules dépendantes de la respiration mitochondriale [9,29-32]. En outre, il a été démontré que le DCA inhibe la croissance d’une série de lignées cellulaires de mélanome, indépendamment du statut BRAF et de la sensibilité au PLX4032 [29]. Pour étudier l’effet combiné de LE135 et du DCA, nous avons appliqué des concentrations inférieures aux valeurs respectives de la CI50 pour chacune des trois lignées cellulaires de mélanome (cf. tableau 1). Malgré un faible effet sur la réduction de la croissance de chaque composé pris individuellement (9-21% pour LE135 et 12-48% pour le DCA), la combinaison a induit une réduction allant jusqu’à 89% (Figure 5E). Ces résultats suggèrent que les effets opposés de LE135 (favorisant la dépendance glycolytique) et du DCA (éloignant les cellules de la glycolyse) peuvent agir en synergie pour inhiber la croissance des mélanomes.
DISCUSSION
L’ATRA et d’autres dérivés de la vitamine A réduisent la croissance cellulaire et induisent l’expression de marqueurs de différenciation dans divers tissus [33, 34]. Nous avons constaté que, dans les mélanocytes humains primaires, l’ATRA induit une régulation transitoire à la hausse de l’axe MITF/PGC1α, ce qui est cohérent avec l’augmentation de la fonction mitochondriale induite par l’ATRA observée dans d’autres types de cellules comme les adipocytes et les hépatocyes [35-37]. Cependant, le traitement à long terme avec de faibles concentrations d’ATRA a entraîné des réductions de la croissance cellulaire et du taux métabolique, déterminées par une glycolyse basale plus faible ainsi qu’une respiration mitochondriale plus faible. Ces changements métaboliques en réponse à l’ATRA reflètent probablement une réponse de différenciation vers l’état non proliférant qui caractérise les mélanocytes résidant dans la peau. Le blocage de la signalisation RARβ dans ces cellules a entraîné une augmentation du taux glycolytique basal et une diminution correspondante du métabolisme oxydatif. L’avantage sélectif de la perte de la fonction de RARβ dans le mélanome, par exemple par hyperméthylation de RARB, pourrait être lié à la transition vers un phénotype plus dépendant de la glycolyse soutenant l’effet Warburg.
Contrairement à la situation dans les mélanocytes primaires, le blocage de la signalisation RARβ dans les cellules de mélanome a conduit à un stress énergétique, comme indiqué par l’activation de l’AMPK. Cette réponse pourrait être le résultat d’une moindre capacité des cellules de mélanome à augmenter la glycolyse par rapport aux mélanocytes. Alors que les mélanocytes ont un niveau glycolytique basal relativement bas et peuvent passer à une activité plus élevée en cas de besoin, les cellules de mélanome sont caractérisées par un taux glycolytique élevé proche de leur capacité maximale. Ainsi, les mélanocytes sont plus flexibles pour s’adapter à l’inhibition de la signalisation RARβ afin de maintenir le niveau d’énergie. Cela indique qu’une fenêtre thérapeutique pertinente existe pour LE135 et d’autres inhibiteurs de RARβ dans des thérapies combinées, par exemple avec le DCA. Comme les effets métaboliques de l’inhibition de BRAF, le DCA fait basculer les cellules cancéreuses glycolytiques vers la respiration mitochondriale [9, 38, 39], mais contrairement au PLX4032, l’effet du DCA n’est pas limité aux mélanomes mutés par BRAF [29]. Dans une étude précédente, nous avons démontré que le changement métabolique induit par le DCA était corrélé à une réduction des niveaux d’ATP, suggérant que le DCA pourrait cibler l’homéostasie bioénergétique des cellules de mélanome [29]. Nous avons constaté ici que la combinaison de LE135 et de DCA atténuait de manière coopérative la croissance des cellules de mélanome exprimant le récepteur RARβ. Le traitement des cellules de mélanome soit avec le DCA soit avec le LE135 pourrait leur donner une fenêtre pour s’adapter aux nouvelles demandes métaboliques, alors que la combinaison des traitements limiterait la flexibilité métabolique et les rendrait incapables de soutenir la production d’énergie nécessaire à la poursuite de la croissance.
Dans de nombreux mélanomes, l’expression de PGC1α est faible en raison de la suppression de MITF par les mutations oncogènes de BRAF [11]. Ce phénotype soutient l’effet Warburg en forçant les cellules à passer à la glycolyse pour la production d’énergie. Le traitement par des inhibiteurs de BRAF restaure l’expression de PGC1α et réoriente le schéma métabolique vers la respiration mitochondriale [11, 40]. L’effet cytotoxique de l’inhibition de BRAF peut être renforcé en raison de la présence de mitochondries dysfonctionnelles dans les cellules de mélanome, ce qui entraîne une production accrue de ROS [13, 41]. Nous avons constaté que l’ATRA augmente la croissance des cellules de mélanome en présence de l’inhibiteur de BRAF PLX4032. Sur la base des connaissances issues d’études précédentes [11, 17, 19, 41] et des résultats présentés ici, nous proposons un modèle intégrant les effets métaboliques du PLX4032 et de l’ATRA et expliquant leur antagonisme (Figure 6). Le modèle suggère un double rôle de l’augmentation de la signalisation RARβ, conduisant à l’activation de PGC1α et de la biogenèse mitochondriale, tout en supprimant l’OCR et la production de ROS par l’induction de p14ARF. Le modèle a été soutenu par les résultats de la présente étude et d’une étude antérieure [17] montrant que le traitement par l’ATRA induit l’expression de p14ARF, et que la reconstitution de cellules de mélanome déficientes en p14ARF avec du p14ARF de type sauvage diminue l’OCR et augmente la sensibilité à l’ATRA. Ainsi, l’ATRA pourrait réduire la sensibilité des mélanomes RARβ-positifs à la PLX4032 en limitant la cytotoxicité de la production de ROS. La vitamine A a été proposée à des fins prophylactiques et thérapeutiques dans de nombreux types de cancer, y compris le mélanome [42]. Bien que la validation clinique fasse défaut, nos résultats plaident contre l’utilisation d’une supplémentation en vitamine A chez les patients atteints de mélanome et traités par des inhibiteurs de BRAF, en raison de l’effet antagoniste potentiel.
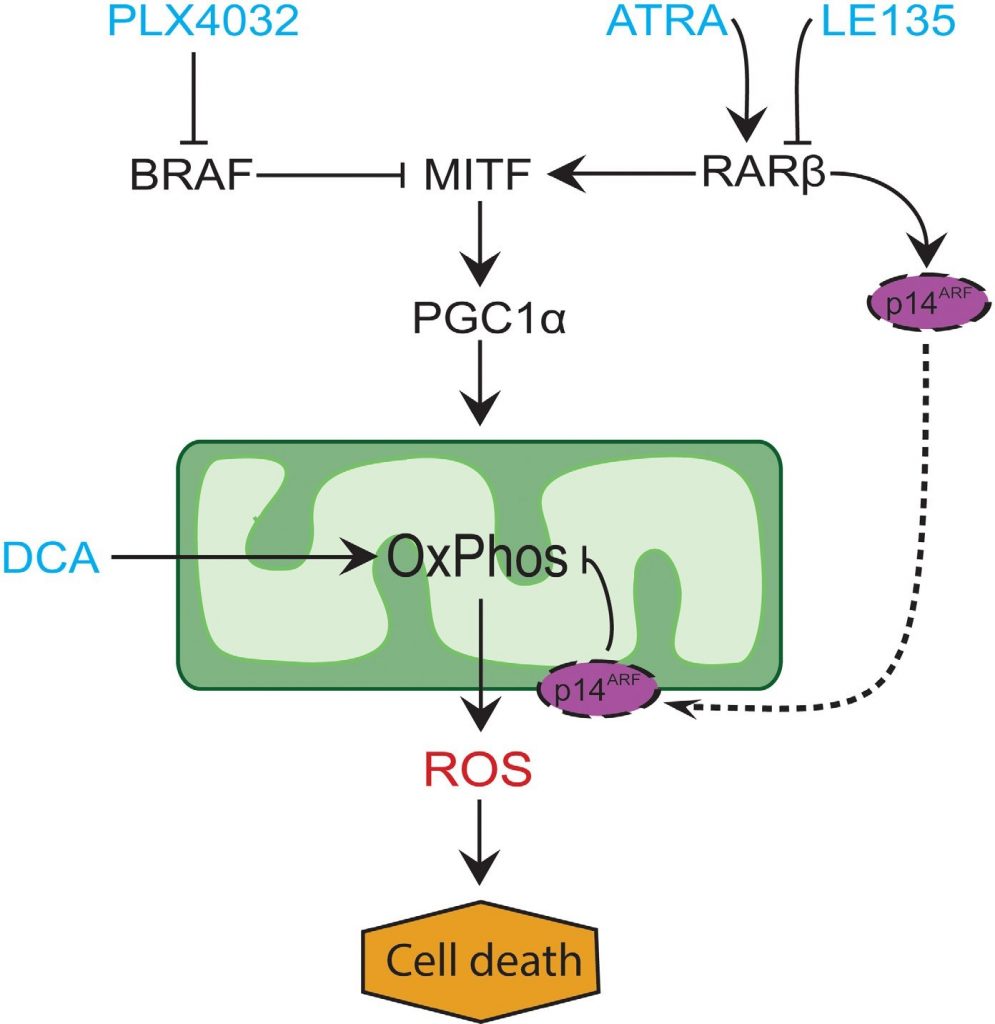
En conclusion, nous avons identifié une nouvelle fonction de la signalisation RARβ dans le métabolisme des cellules mélanocytaires et des mélanomes, qui pourrait avoir des implications cliniques. La capacité de RARβ à activer la voie MITF-PGC1α, et potentiellement une réduction de l’activité respiratoire mitochondriale dépendante de p14ARF, affecte négativement la réponse thérapeutique à l’inhibition de BRAF. Cependant, le blocage de la signalisation RARβ favorise la dépendance glycolytique dans les cellules de mélanome et améliore l’effet du DCA, ce qui pourrait potentiellement être exploité sur le plan thérapeutique.
MATÉRIEL ET MÉTHODES
Réactifs
Le dichloroacétate de sodium (DCA), le 2-désoxy-D-glucose (2-DG), l’acide rétinoïque all-trans (ATRA) et le LE135 ont été achetés chez Sigma-Aldrich. Le DCA et le 2-DG ont été dissous dans du dH2Ojusqu’à une concentration de travail de 1 M. L’ATRA et le LE135 ont été dissous dans du DMSO jusqu’à une concentration de travail de 0,1 M. Le PLX4032 (vemurafenib) a été acheté auprès de Selleck Chemicals et dissous dans du DMSO jusqu’à une concentration de travail de 0,05 M.
Culture cellulaire
Des lignées cellulaires de mélanome ont été obtenues à partir de la base de données européenne des lignées tumorales (ESTDAB, ED) [43]. Le statut de ces lignées cellulaires en ce qui concerne les mutations BRAF et la méthylation du promoteur RARB a été décrit précédemment [17]. Des mélanocytes épidermiques humains primaires (néonataux) provenant de tissus légèrement pigmentés (HEMn-LP ; appelés mélanocytes) ont été achetés chez Invitrogen (C0025C). Des mélanocytes provenant de trois individus différents ont été utilisés pour les expériences (Lot n° 200706893 de mars 2015 et nov. 2016 ; Lot n° 1583282 de février 2017). Les lignées cellulaires de mélanome ont été cultivées à 37°C sous 5% deCO2 dans un milieu RPMI-1640 complété par 10% de sérum bovin fœtal. Les cellules HEMn-LP ont été cultivées dans les mêmes conditions dans un milieu 254CF complété par 1 % de supplément de croissance des mélanocytes humains (HMGS) incluant du phorbol 12-myristate 13-acétate (PMA). Pour les montages expérimentaux, les cellules HEMn-LP ont été cultivées dans un milieu supplémenté avec HMGS-2 (sans PMA). Tous les milieux et suppléments ont été achetés chez Invitrogen.
Analyse de la viabilité cellulaire
Untest au cristal violet a été appliqué pour évaluer l’effet des composés étudiés sur la viabilité cellulaire. Les cellules ont été ensemencées en double et traitées avec les composés concernés ou le véhicule de contrôle pendant 6 jours. Le milieu et les composés de traitement ont été remplacés toutes les 48 h. Les expériences ont été répétées trois fois indépendamment. Après la période de traitement, le milieu et les cellules non attachées ont été retirés, et les cellules restantes ont été lavées dans du PBS et fixées avec du glutaraldéhyde pendant 15 minutes. Les cellules fixées ont été incubées avec une solution de cristal violet (0,1 % de cristal violet, 20 % de CH3OH) pendant 1 h. La quantité de colorant absorbée par la monocouche, proportionnelle au nombre de cellules viables attachées au fond du puits, a été quantifiée en extrayant la couleur avec de l’acide acétique à 10 % et en mesurant l’absorbance à une longueur d’onde de 595 nm. La viabilité relative après traitement par ATRA, LE135 ou PLX4032 a été utilisée pour déterminer la concentration inhibitrice semi-maximale (IC50). En traçant la courbe dose-réponse, la valeur de la CI50 a été estimée comme étant la concentration au point de viabilité cellulaire de 50 %.
Purification de l’ADN et de l’ARN
L’ADN pour la quantification de l’ADNmt et l’ARN pour la synthèse de l’ADNc ont été purifiés simultanément avec le kit AllPrep DNA/RNA/Protein mini (Qiagen) selon le protocole fourni.
Analyse de l’expression
La synthèse d’ADNc a été réalisée avec le qScript™ XLT cDNA SuperMix (Quanta Bioscience). L’expression génétique de PGC1α, MITF, RARβ, p14ARF, UCP2, ATP5g1, COX5A et NDUFS3 a été déterminée par PCR quantitative en temps réel sur le LightCycler 2.0 de Roche en utilisant le kit LightCycler FastStart DNA MasterPLUS SYBR Green I (Roche). Les amorces sont énumérées dans le tableau supplémentaire 1.
| Gène | Amorce directe | Amorce inverse |
| PGC1α* | GTAAATCTGCGGGATGATGG | AATTGCTTGCGTCCACAAA |
| MITF** | CCGTCTCTCACTGGATTGGT | TACTTGGTGGGGTTTTCGAG |
| p14ARF*** | CCCTCGTGCTGATGCTACTGA | CATGACCTGGTCTTAGGAAGC |
| RARβ*** | TCCTGGATTTCTACACTGCG | AAGCAGGGTTTGTACACTCG |
| UCP2 | AAGACCATTGCCCGAGG | TTGGCTTTCAGGAGGGCAT |
| ATP5g1* | ATCATTGGCTATGCCAGGAA | ATGGCGAAGGATGAGGA |
| COX5A* | GGGAATTGCGTAAAGGGATAA | TCCTGCTTTGTCCTTAACAACC |
| NDUFS3* | GCTGACGCCCATTGAGTCTG | GGAACTCTTGGGCCAACTCC |
| RPLP0 | ACTAAAATCTCCAGGGGCACC | ATGACCAGCCCAAAGGAGAA |
*Séquences d’amorces publiées par Vazquez et al. 2013 [22].
**Séquences d’amorces publiées par Haq et al. 2014 [11].
***Séquences d’amorces publiées par Dahl et al. 2013 [17].
Immunoblotting
Les échantillonsont été préparés à partir de flacons de culture cellulaire avec du tampon de lyse (SLB) complété par du β-mercaptoéthanol incolore (BPB), du Phospho-Stop et un inhibiteur de protéase (Thermo Fisher Scientific). Les lysats cellulaires ont été nettoyés par centrifugation à 20 000 rpm pendant 3 min. La concentration en protéines a été mesurée à l’aide du kit de dosage des protéines Qubit (Thermo Fisher Scientific), et 50 μg de protéines de chaque échantillon ont été chargés sur un gel SDS, 4-12% Bis-Tris NuPage à 10 puits (Invitrogen). Les protéines ont ensuite été séparées à 80 V pendant 30 min, puis à 110 V jusqu’à la fin. Le transfert a été effectué avec une unité de transfert semi-sec sur une membrane de nitrocellulose ECL à 3,3 mA/1 cm2/1 h/gel. Ensuite, la membrane a été colorée avec du Ponceau. La membrane a été bloquée dans du lait à 5% pendant 1 h, puis lavée deux fois pendant 5 min avec du TBST et colorée avec des anticorps anti-AMPK ou anti-p-AMPK (Thr172) (Cell Signaling ; 1:2000) dans 5% de BSA à 4°C et avec un anticorps anti-cyclophiline A (Cell Signaling ; 1:5000) comme contrôle de charge. Après trois cycles de lavage de 10 min avec TBST, la membrane a été colorée avec l’anticorps secondaire (anti-lapin ; DakoCytomation ; 1:2000) pendant 1 h à température ambiante, suivi de 3 autres cycles de lavage. Les protéines ont été visualisées en utilisant le substrat de Western Blotting ECL Plus (Thermo Fisher Scientific) 1:1 pendant 2 à 3 minutes.
Analyse métabolique
L’analyse métabolique a été réalisée sur des lignées cellulaires de mélanome et des mélanocytes à l’aide d’un analyseur Seahorse XFe96 (Seahorse Bioscience, Billerica, MA), qui effectue des mesures en temps réel du taux d’acidification extracellulaire (ECAR) et du taux de consommation d’oxygène (OCR). Les cellules ont été ensemencées à 20 000 par puits dans des microplaques de culture cellulaire Seahorse 24 heures avant d’effectuer les mesures. Les changements dans l’activité basale et la capacité des systèmes énergétiques mitochondriaux et glycolytiques ont été déterminés à l’aide du Mito Stress Test Kit et du Glycolysis Stress Test Kit (Agilent Technologies). Les tests ont été réalisés conformément aux protocoles fournis. Le test de stress mitotique a été réalisé dans le milieu de culture habituel, alors que dans le test de stress glycolytique, le milieu a été remplacé par le milieu Seahorse XF Base complété par de la L-glutamine (2 mM), pH ajusté à 7,4, 1 h avant les mesures. Pour les expositions plus longues (>24 h), les cellules ont été traitées dans des flacons de culture avant d’être ensemencées. Tous les résultats ont été normalisés par rapport au nombre de cellules ensemencées, car les concentrations d’ATRA et de LE135 utilisées n’ont pas affecté la croissance cellulaire pendant 24 h. Le protocole d’exécution des tests dans l’appareil Seahorse comprenait des cycles de 3 min de mélange/3 min de mesure. Trois expériences indépendantes ont été réalisées avec 6 répliques de chaque échantillon.
Transfection
La lignée cellulaire de mélanome ED-007 a été transfectée avec les vecteurs d’expression pEGFP (contrôle) ou pEGFP-p14ARF (2 μg de vecteur/2 ×106 cellules), contenant tous deux la GFP comme gène rapporteur. Les constructions ont été obtenues comme décrit précédemment [19]. La transfection a été réalisée en utilisant la technologie de nucléofection Amaxa, le tampon V, le programme T-020, en suivant le protocole recommandé par le fabricant. La réussite de la transfection a été vérifiée visuellement. Les clones stables ont été sélectionnés en utilisant 400 μg/ml de G418 (généine ; Thermo Fisher Scientific). Dans le montage expérimental, les cellules ont été ensemencées sans G418.
Quantification de l’ADN mitochondrial
L’ADN mitochondrial a été quantifié par réaction en chaîne par polymérase numérique en gouttelettes (ddPCR) à l’aide du système QX200 (BioRad Laboratories, Hercules, CA, USA). Environ 0,5 ng d’ADN a été utilisé pour chaque réaction. Le nombre de copies mitochondriales a été déterminé en calculant le rapport entre un site d’ADN mitochondrial (mtMinArc) et un locus nucléaire à copie unique (β2m), comme décrit par Phillips et al [44]. Les amorces, les sondes et les conditions expérimentales sont énumérées dans le tableau supplémentaire 2.
| mtMinArc | β2m | |
| Amorce directe | CTAAATAGCCCACACGTTCCC | GCTGGGTAGCTCTAAACAATGTATTCA |
| Amorce inverse* | AGAGCTCCCGTGAGTGGTTA | CCATGTACTAACAAATGTCTAAAATGGT |
| Sonde* | 6FAM-CATCACGATGGATCACAGGT(NFQ) | VIC-CAGCAGCCTATTCTGC(NFQ) |
| Conc. d’amorces | 75 nM | 500 nM |
| Temp. de recuit | 50°C | 52°C |
| Nombre de cycles | 40 | 40 |
*Séquences des amorces et des sondes publiées par Phillips et al. [44].
Analyse statistique
Les différences entre des ensembles de données indépendants ont été déterminées à l’aide du test t de Student. L’ANOVA à échantillons appariés à une voie a été utilisée pour l’analyse statistique de la variance entre les différents traitements. Le test de comparaison multiple de Tukey (HSD, honest significance difference) a été utilisé pour déterminer la signification statistique.
Contributions des auteurs
CA et PG ont planifié et organisé l’étude. CA a réalisé la majorité des expériences et le traitement des données, y compris la culture cellulaire, les protocoles de traitement, l’analyse métabolique et les statistiques. CD a planifié et réalisé la transfection EGFP-p14ARF et les mesures de l’ADNmt et a participé à l’interprétation des résultats. AA a réalisé et optimisé les protocoles d’immunoblotting et de PCR quantitative. AC a réalisé les essais de culture cellulaire. CA et PG ont rédigé le manuscrit avec les contributions et les révisions de CD, AA et AC. Le manuscrit final a été lu et approuvé par tous les auteurs.
CONFLITS D’INTÉRÊTS
Les auteurs déclarent qu’ils n’ont pas de conflits d’intérêts.
FINANCEMENT
Cette étude a été soutenue par la Société danoise du cancer
RÉFÉRENCES
1 Taux de survie pour le cancer de la peau mélanome, par stade. (cancer.org : American Cancer Society).2 Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, Chodon T, Nelson SF, McArthur G, et al. Melanomas acquire resistance to B-RAF (V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010 ; 468 : 973-7. https://doi.org/10.1038/nature09626.
3 Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP, Barretina J, Caponigro G, Hieronymus H, Murray RR, et al. COT conduit la résistance à l’inhibition de RAF par la réactivation de la voie MAP kinase. Nature. 2010 ; 468 : 968-72. https://doi.org/10.1038/nature09627.
4 Miller AJ, Mihm MC Jr. Melanoma. N Engl J Med. 2006 ; 355 : 51-65. https://doi.org/10.1056/NEJMra052166.
5 Abildgaard C, Guldberg P. Molecular drivers of cellular metabolic reprogramming in melanoma. Trends Mol Med. 2015 ; 21 : 164-71. https://doi.org/10.1016/j.molmed.2014.12.007.
6 Ratnikov BI, Scott DA, Osterman AL, Smith JW, Ronai ZA. Le recâblage métabolique dans le mélanome. Oncogene. 2017 ; 36 : 147-57. https://doi.org/10.1038/onc.2016.198.
7 Warburg O, Wind F, Negelein E. Le métabolisme des tumeurs dans l’organisme. J Gen Physiol. 1927 ; 8 : 519-30.
8 Hall A, Meyle KD, Lange MK, Klima M, Sanderhoff M, Dahl C, Abildgaard C, Thorup K, Moghimi SM, Jensen PB, Bartek J, Guldberg P, Christensen C. La phosphorylation oxydative dysfonctionnelle rend les cellules de mélanome malin dépendantes de la glycolyse dirigée par l’oncogène (V600E) BRAF. Oncotarget. 2013 ; 4 : 584-99. https://doi.org/10.18632/oncotarget.965.
9 Parmenter TJ, Kleinschmidt M, Kinross KM, Bond ST, Li J, Kaadige MR, Rao A, Sheppard KE, Hugo W, Pupo GM, Pearson RB, McGee SL, Long GV, et al. La réponse du mélanome mutant BRAF à l’inhibition de BRAF est médiée par un réseau de régulateurs transcriptionnels de la glycolyse. Cancer Discov. 2014 ; 4 : 423-33. https://doi.org/10.1158/2159-8290.CD-13-0440.
10 McArthur GA, Puzanov I, Amaravadi R, Ribas A, Chapman P, Kim KB, Sosman JA, Lee RJ, Nolop K, Flaherty KT, Callahan J, Hicks RJ. Réponses marquées, homogènes et précoces de la tomographie par émission de positons au [18F]fluorodésoxyglucose au vemurafenib dans le mélanome avancé BRAF-mutant. J Clin Oncol. 2012 ; 30 : 1628-34. https://doi.org/10.1200/JCO.2011.39.1938.
11 Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, Frederick DT, Hurley AD, Nellore A, Kung AL, Wargo JA, Song JS, Fisher DE, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell. 2013 ; 23 : 302-15. https://doi.org/10.1016/j.ccr.2013.02.003.
12 Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, et al. Amélioration de la survie avec le vemurafenib dans le mélanome avec mutation BRAF V600E. N Engl J Med. 2011 ; 364 : 2507-16. https://doi.org/10.1056/NEJMoa1103782.
13 Corazao-Rozas P, Guerreschi P, Jendoubi M, Andre F, Jonneaux A, Scalbert C, Garcon G, Malet-Martino M, Balayssac S, Rocchi S, Savina A, Formstecher P, Mortier L, et al. Le stress oxydatif mitochondrial est le talon d’Achille des cellules de mélanome résistantes à l’inhibiteur de la mutation de Braf. Oncotarget. 2013 ; 4 : 1986-98. https://doi.org/10.18632/oncotarget.1420.
14 Zhang G, Frederick DT, Wu L, Wei Z, Krepler C, Srinivasan S, Chae YC, Xu X, Choi H, Dimwamwa E, Ope O, Shannan B, Basu D, et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J Clin Invest. 2016 ; 126 : 1834-56. https://doi.org/10.1172/JCI82661.
15 Livingstone E, Swann S, Lilla C, Schadendorf D, Roesch A. Combiner l’inhibition de BRAF(V) (600E) avec des modulateurs du métabolisme bioénergétique mitochondrial pour surmonter la résistance aux médicaments dans le mélanome métastatique. Exp Dermatol. 2015 ; 24 : 709-10. https://doi.org/10.1111/exd.12718.
16 Hoon DS, Spugnardi M, Kuo C, Huang SK, Morton DL, Taback B. Profiling epigenetic inactivation of tumor suppressor genes in tumors and plasma from cutaneous melanoma patients. Oncogene. 2004 ; 23 : 4014-22. https://doi.org/10.1038/sj.onc.1207505.
17 Dahl C, Christensen C, Jonsson G, Lorentzen A, Skjodt ML, Borg A, Pawelec G, Guldberg P. Une analyse d’exclusivité mutuelle des facteurs génétiques et épigénétiques dans le mélanome identifie un lien entre p14 ARF et la signalisation RARbeta. Mol Cancer Res. 2013 ; 11 : 1166-78. https://doi.org/10.1158/1541-7786.MCR-13-0006.
18 Lotan R, Lotan D. Amélioration de l’expression mélanotique dans les cellules de mélanome de souris en culture par les rétinoïdes. J Cell Physiol. 1981 ; 106 : 179-89. https://doi.org/10.1002/jcp.1041060203.
19 Christensen C, Bartkova J, Mistrik M, Hall A, Lange MK, Ralfkiaer U, Bartek J, Guldberg P. A short acidic motif in ARF guards against mitochondrial dysfunction and melanoma susceptibility. Nat Commun. 2014 ; 5 : 5348. https://doi.org/10.1038/ncomms6348.
20 Baldea I, Costin GE, Shellman Y, Kechris K, Olteanu ED, Filip A, Cosgarea MR, Norris DA, Birlea SA. Effets biphasiques pro-mélanogènes et pro-apoptotiques de l’acide all-trans-rétinoïque (ATRA) sur les mélanocytes humains : étude du cours du temps. J Dermatol Sci. 2013 ; 72 : 168-76. https://doi.org/10.1016/j.jdermsci.2013.06.004.
21 Kawakami T, Ohgushi A, Hirobe T, Soma Y. Analyse des effets de l’acide all-trans rétinoïque sur les mélanocytes et les mélanoblastes humains in vitro. J Dermatol. 2017 ; 44 : 93-4. https://doi.org/10.1111/1346-8138.13477.
22 Vazquez F, Lim JH, Chim H, Bhalla K, Girnun G, Pierce K, Clish CB, Granter SR, Widlund HR, Spiegelman BM, Puigserver P. L’expression de PGC1alpha définit un sous-ensemble de tumeurs de mélanome humain avec une capacité mitochondriale accrue et une résistance au stress oxydatif. Cancer Cell. 2013 ; 23 : 287-301. https://doi.org/10.1016/j.ccr.2012.11.020.
23 Donadelli M, Dando I, Fiorini C, Palmieri M. UCP2, une protéine mitochondriale régulée à de multiples niveaux. Cell Mol Life Sci. 2014 ; 71 : 1171-90. https://doi.org/10.1007/s00018-013-1407-0.
24 Oberkofler H, Klein K, Felder TK, Krempler F, Patsch W. Role of peroxisome proliferator-activated receptor-gamma coactivator-1alpha in the transcriptional regulation of the human uncoupling protein 2 gene in INS-1E cells. Endocrinologie. 2006 ; 147 : 966-76. https://doi.org/10.1210/en.2005-0817.
25 Arigony AL, de Oliveira IM, Machado M, Bordin DL, Bergter L, Pra D, Henriques JA. L’influence des micronutriments en culture cellulaire : une réflexion sur la viabilité et la stabilité génomique. Biomed Res Int. 2013 ; 2013 : 597282. https://doi.org/10.1155/2013/597282.
26 Li Y, Hashimoto Y, Agadir A, Kagechika H, Zhang X. Identification d’une nouvelle classe d’antagonistes rétinoïdes sélectifs du récepteur bêta de l’acide rétinoïque et leurs effets inhibiteurs sur l’activité AP-1 et l’apoptose induite par l’acide rétinoïque dans les cellules cancéreuses du sein humain. J Biol Chem. 1999 ; 274 : 15360-6.
27 de The H, Marchio A, Tiollais P, Dejean A. Differential expression and ligand regulation of the retinoic acid receptor alpha and beta genes. EMBO J. 1989 ; 8 : 429-33.
28 Hardie DG, Hawley SA. AMP-activated protein kinase : the energy charge hypothesis revisited. Bioessays. 2001 ; 23 : 1112-9. https://doi.org/10.1002/bies.10009.
29 potentialise leur réponse à l’inhibition de BRAFV600E. J Transl Med. 2014 ; 12 : 247. https://doi.org/10.1186/s12967-014-0247-5.
30 Populo H, Caldas R, Lopes JM, Pardal J, Maximo V, Soares P. La surexpression de la pyruvate déshydrogénase kinase soutient le dichloroacétate comme candidat pour la thérapie du mélanome cutané. Expert Opin Ther Targets. 2015 ; 19 : 733-45. https://doi.org/10.1517/14728222.2015.1045416.
31 Kluza J, Corazao-Rozas P, Touil Y, Jendoubi M, Maire C, Guerreschi P, Jonneaux A, Ballot C, Balayssac S, Valable S, Corroyer-Dulmont A, Bernaudin M, Malet-Martino M, et al. L’inactivation de l’axe de signalisation HIF-1alpha/PDK3 conduit le mélanome vers le métabolisme oxydatif mitochondrial et potentialise l’activité thérapeutique des pro-oxydants. Cancer Res. 2012 ; 72 : 5035-47. https://doi.org/10.1158/0008-5472.CAN-12-0979.
32 Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, van der Burg SH, Verdegaal EM, Cascante M, Shlomi T, Gottlieb E, Peeper DS. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature. 2013 ; 498 : 109-12. https://doi.org/10.1038/nature12154.
33 Murholm M, Isidor MS, Basse AL, Winther S, Sorensen C, Skovgaard-Petersen J, Nielsen MM, Hansen AS, Quistorff B, Hansen JB. L’acide rétinoïque a des effets différents sur l’expression d’UCP1 dans les adipocytes de souris et d’humains. BMC Cell Biol. 2013 ; 14 : 41. https://doi.org/10.1186/1471-2121-14-41.
34 Jin W, Xu YP, Yang AH, Xing YQ. Induction et différenciation in vitro de cellules souches mésenchymateuses de cordon ombilical en cellules de type neurones par l’acide rétinoïque all-trans. Int J Ophthalmol. 2015 ; 8 : 250-6. https://doi.org/10.3980/j.issn.2222-3959.2015.02.07.
35 Tourniaire F, Musinovic H, Gouranton E, Astier J, Marcotorchino J, Arreguin A, Bernot D, Palou A, Bonet ML, Ribot J, Landrier JF. L’acide rétinoïque all-trans induit la phosphorylation oxydative et la biogenèse des mitochondries dans les adipocytes. J Lipid Res. 2015 ; 56 : 1100-9. https://doi.org/10.1194/jlr.M053652.
36 Tripathy S, Chapman JD, Han CY, Hogarth CA, Arnold SL, Onken J, Kent T, Goodlett DR, Isoherranen N. L’acide all-trans rétinoïque améliore la fonction mitochondriale dans des modèles de foie humain. Mol Pharmacol. 2016 ; 89 : 560-74. https://doi.org/10.1124/mol.116.103697.
37 Watabe H, Soma Y, Ito M, Kawa Y, Mizoguchi M. L’acide all-trans rétinoïque induit la différenciation et l’apoptose des précurseurs des mélanocytes murins avec induction du facteur de transcription associé à la microphtalmie. J Invest Dermatol. 2002 ; 118 : 35-42. https://doi.org/10.1046/j.0022-202x.2001.01614.x.
38 De Preter G, Neveu MA, Danhier P, Brisson L, Payen VL, Porporato PE, Jordan BF, Sonveaux P, Gallez B. Inhibition of the pentose phosphate pathway by dichloroacetate unravels a missing link between aerobic glycolysis and cancer cell proliferation. Oncotarget. 2016 ; 7 : 2910-20. https://doi.org/10.18632/oncotarget.6272.
39 Michelakis ED, Webster L, Mackey JR. Le dichloroacétate (DCA) comme une thérapie potentielle ciblant le métabolisme pour le cancer. Br J Cancer. 2008 ; 99 : 989-94. https://doi.org/10.1038/sj.bjc.6604554.
40 Corazao-Rozas P, Guerreschi P, Andre F, Gabert PE, Lancel S, Dekiouk S, Fontaine D, Tardivel M, Savina A, Quesnel B, Mortier L, Marchetti P, Kluza J. Mitochondrial oxidative phosphorylation controls cancer cell’s life and death decisions upon exposure to MAPK inhibitors. Oncotarget. 2016 ; 7 : 39473-85. https://doi.org/10.18632/oncotarget.7790.
41 Bauer D, Werth F, Nguyen HA, Kiecker F, Eberle J. Rôle critique des espèces réactives de l’oxygène (ROS) pour l’amélioration synergique de l’apoptose par le vemurafenib et l’inhibiteur de canal de potassium TRAM-34 dans les cellules de mélanome. Cell Death Dis. 2017 ; 8 : e2594. https://doi.org/10.1038/cddis.2017.6.
42 Chen MC, Hsu SL, Lin H, Yang TY. L’acide rétinoïque et le traitement du cancer. Biomedicine (Taipei). 2014 ; 4 : 22. https://doi.org/10.7603/s40681-014-0022-1.
43 Robinson J, Roberts CH, Dodi IA, Madrigal JA, Pawelec G, Wedel L, Marsh SG. La base de données européenne de lignées tumorales consultable. Cancer Immunol Immunother. 2009 ; 58 : 1501-6. https://doi.org/10.1007/s00262-008-0656-5.
44 Phillips NR, Sprouse ML, Roby RK. Quantification simultanée du nombre de copies d’ADN mitochondrial et du rapport de délétion : un test PCR en temps réel multiplex. Sci Rep. 2014 ; 4 : 3887. https://doi.org/10.1038/srep03887.