Ronen Shavit1, Maya Ilouze, Tali Feinberg, Yaacov Richard Lawrence, Yossi Tzur, Nir Peled
1 Thoracic Cancer Research and Detection Center, Sheba Medical Center Tel Hashomer, Ramat-Gan, 52621, POB 244, Israël.
e-mail : [email protected]
URL :http://medicine.mytau.org/peled/
Y. R.Lawrence
Centre de recherche translationnelle en radio-oncologie, Sheba Medical Center, Ramat-Gan, Israël
M. Ilouze : N. Peled
Service du cancer thoracique, Davidoff Cancer Center, Rabin Medical Center, Petach Tikva, Israël
Correspondance : [email protected]
Accepté : 10 décembre 2014
Publié : 7 janvier 2015
Résumé
Introduction : Le cancer du poumon est la principale cause de décès par cancer. La radiothérapie joue un rôle clé dans son traitement. Les rayonnements ionisants induisent la mort cellulaire par des aberrations chromosomiques, qui déclenchent la catastrophe mitotique et l’apoptose. Cependant, de nombreux patients atteints de cancer du poumon présentent une résistance aux radiations. Le dichloroacétate (DCA) est une petite molécule qui peut favoriser l’activation mitochondriale en augmentant l’afflux de pyruvate. Nous avons testé ici si le DCA pouvait augmenter la sensibilité des cellules de cancer du poumon non à petites cellules (NSCLC) aux radiations par ce mécanisme.
Méthodes : Deux lignées cellulaires représentatives du cancer du poumon non à petites cellules (A549 et H1299) ont été testées pour leur sensibilité aux rayonnements avec et sans exposition préalable au DCA. L’efficacité du traitement a été évaluée à l’aide d’un test de survie clonogénique. Un analyseur de flux extracellulaire a été utilisé pour évaluer l’effet du DCA sur la consommation d’oxygène cellulaire, un marqueur de substitution de l’activité mitochondriale.
Résultats : Nous avons constaté que le DCA augmente le taux de consommation d’oxygène des cellules A549 et H1299 de 60 % (p = 0,0037) et de 20 % (p = 0,0039), respectivement. La pré-exposition au DCA une heure avant l’irradiation a multiplié le taux de mort cytotoxique par 4 dans les cellules A549 (55 à 13%, p = 0,004) et par 2 dans les cellules H1299 (35 à 17%, p = 0,28) respectivement, par rapport à l’irradiation seule.
Conclusion : L’induction mitochondriale par le DCA peut servir de radio-sensibilisateur dans le cancer du poumon non à petites cellules.
Mots clés : NSCLC ; DCA ; Mitochondries ; Rayonnement ; Radio-sensibilisateur ; Effet Warburg
© Société internationale d’oncologie cellulaire 2015
INTRODUCTION
Le cancer du poumon est la principale cause de décès liés au cancer aux États-Unis, avec un taux de survie global à 5 ans, tous stades confondus, de ~17 % [1-4]. La radiothérapie (RT) joue un rôle important dans la prise en charge clinique des patients atteints de cancer du poumon, en particulier ceux qui présentent une maladie de stade IIIB et qui sont candidats à une chimio-radiothérapie définitive. En outre, la RT peut être appliquée en tant que thérapie néo-adjuvante ou adjuvante au stade IIIA, ou de manière ablative lorsque la radiothérapie stéréotaxique du corps (SBRT) est appliquée. La pneumonie radio-induite (RIP) est le facteur limitant lors du traitement des patients par RT. Afin de minimiser la PIR, les oncologues visent à maintenir le V20 (c’est-à-dire le pourcentage du volume pulmonaire recevant une dose de rayonnement de ≥20 Gy) en dessous de 22 % [5]. Les radiosensibilisateurs peuvent augmenter l’efficacité cytotoxique du rayonnement, améliorant ainsi potentiellement les taux de guérison sans augmenter le V20.
La RT tue les cellules en provoquant des cassures double brin de l’ADN. Les cassures d’ADN non réparées entraînent des aberrations chromosomiques qui, à leur tour, conduisent à la « catastrophe mitotique » – un mode de mort cellulaire qui résulte de l’entrée prématurée ou inappropriée des cellules en mitose [6,7]. En outre, les rayonnements peuvent affecter directement les membranes et les organites des cellules. Bien que ces modifications soient encore mal comprises, elles peuvent entraîner des changements dans la transduction des signaux, l’expression des gènes, la stabilité des protéines, les états d’oxydoréduction cellulaires et la régulation du cycle cellulaire, qui peuvent tous conduire à l’apoptose [8]. En outre, les rayonnements peuvent induire la production d’espèces réactives de l’oxygène (ROS) mitochondriales, accompagnée d’une régulation à la hausse des fonctions de la chaîne de transport d’électrons mitochondriale, augmentant ainsi le potentiel de la membrane mitochondriale, la respiration mitochondriale et la production d’ATP mitochondriale [9,10]. Cependant, les altérations génétiques limitent la capacité des cellules cancéreuses à subir l’apoptose, ce qui suggère que les micro-environnements tumoraux hypoxiques peuvent exercer une pression sélective vers un phénotype cancéreux résistant à l’apoptose et, par conséquent, une résistance à la RT [11]. En raison de ce phénotype résistant à la RT et des effets secondaires importants causés par la RT elle-même, il est d’une importance cruciale d’explorer les moyens de radiosensibiliser les cellules cancéreuses du poumon afin de diminuer les doses de radiation et d’améliorer les réponses à la thérapie.
De nombreuses cellules tumorales présentent des niveaux élevés d’absorption du glucose et des niveaux réduits de phosphorylation oxydative. Cette hyperglycolyse paradoxale et ce manque d’activité mitochondriale en présence d’oxygène sont connus sous le nom d’effet Warburg (voir la figure 1) [12]. Le métabolisme unique de la plupart des tumeurs solides provient du remodelage des fonctions mitochondriales pour produire un phénotype glycolytique et une forte résistance à l’apoptose [13]. De plus en plus d’éléments indiquent que les mitochondries pourraient être les principales cibles des traitements anticancéreux [14-17]. Le remodelage spécifique du cancer peut être inversé par une petite molécule appelée dichloroacétate (DCA) [18], qui inhibe la pyruvate déshydrogénase kinase (PDK), augmentant ainsi l’afflux de pyruvate dans la mitochondrie et favorisant son oxydation par l’activité mitochondriale plutôt que par la glycolyse dans divers types de cancer (voir la figure 1) [13,18]. Une augmentation de l’activité mitochondriale entraîne une augmentation de la quantité et de l’ampleur de la libération de radicaux libres (ROS) dans la cellule, ce qui conduit à une augmentation de l’activité apoptotique cellulaire [13].
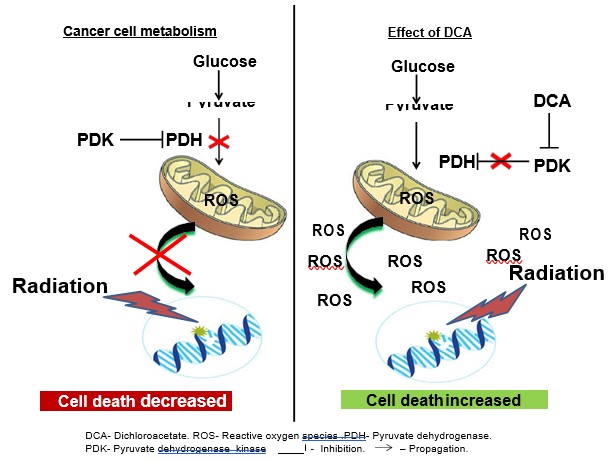
Nous avons étudié ici l’effet radiosensible potentiel du DCA sur les cellules de NSCLC. Nos résultats indiquent que l’activation mitochondriale par le DCA augmente la sensibilité aux radiations dans les cellules A549 et H1299 dérivées du NSCLC.
Matériel et méthodes
Cultures cellulaires
Deux lignées cellulaires de cancer du poumon non à petites cellules (NSCLC) ont été utilisées dans cette étude : A549, dérivée d’un adénocarcinome humain et H1299, dérivée d’un carcinome humain à grandes cellules. Les deux lignées cellulaires ont été achetées auprès de l’ATCC. Les cellules ont été cultivées dans un milieu RPMI-1640 complété par 10 % de sérum bovin fœtal (FBS), 1 % de pénicilline/streptomycine et 1 % de glutamate à 37 °C dans une atmosphère humidifiée à 5 % deCO2.
Conditions de traitement
Ledichloroacétate(DCA, Sigma 34795) a été ajouté aux milieux de culture pendant 1 h avant l’irradiation à 10 et 20 mM dans le cas des cellules H1299 et à 40 et 60 mM dans le cas des cellules A549, selon les valeurs respectives de la CI50. Les cellules ont été irradiées avec un irradiateur Kimtron Polaris à un débit de dose de 0,9 Gy/min à température ambiante.
Test de survie clonogénique
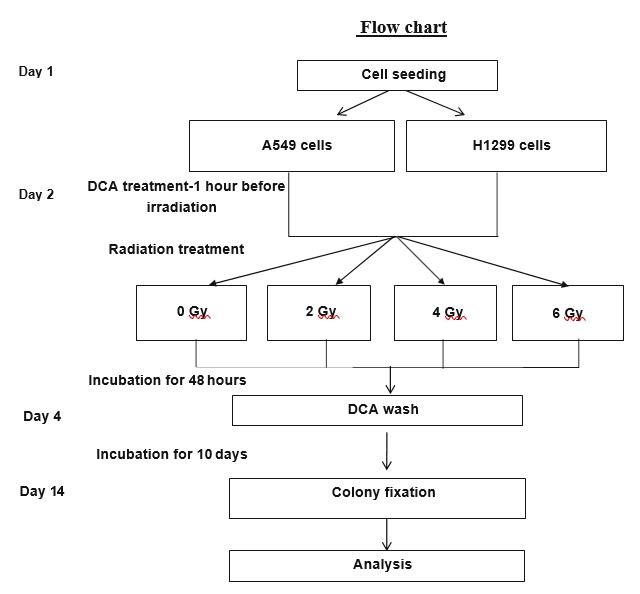
Le test de survie clonogénique que nous avons utilisé a été décrit précédemment [19]. En bref, les cellules ont été détachées à l’aide de trypsine, mises en suspension dans un milieu de culture complet, comptées et ensemencées dans des boîtes de 3 ml à une densité de 200 cellules par boîte, puis on les a laissées se fixer et se stabiliser pendant la nuit. Le lendemain, les groupes testés ont été traités au DCA (ou au contrôle) 1 h avant l’irradiation, aux concentrations indiquées. Les cellules ont été irradiées à des doses de 0, 2, 4 et 6 Gy. Après une incubation de 48 heures, les milieux de culture ont été retirés et remplacés par des milieux de culture frais sans DCA (tous les groupes). Les cellules ont été incubées pendant 10 jours supplémentaires pour permettre la formation de colonies. Ensuite, les colonies ont été fixées et colorées avec du cristal violet à 0,5 % dans de l’éthanol à 96 %. Les colonies contenant au moins 50 cellules viables ont été comptées (Fig. 2). Chaque expérience a été réalisée en double et la moyenne et l’écart-type ont été calculés à partir de 3 expériences indépendantes.
Évaluation de la synergie
Afin d’évaluer si les schémas de traitement présentent des effets synergiques, des graphiques des moyennes avec les écarts types des taux de survie par rapport aux doses de rayonnement ont été réalisés. Les pourcentages de survie ont été calculés, à la fois pour les groupes traités et non traités au DCA, par rapport à ceux des cellules non irradiées :
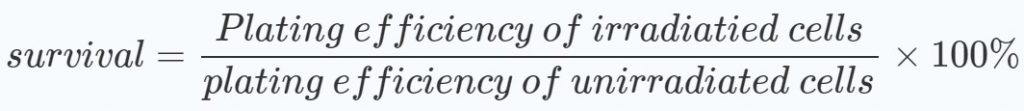
où :
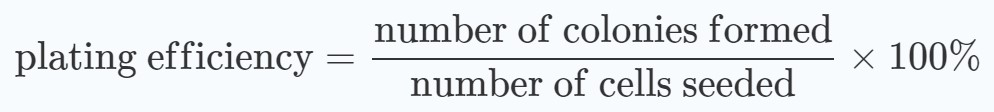
Plus précisément, la formation de colonies des groupes traités au DCA et non traités au DCA sans radiothérapie a été normalisée à 100 %. Les effets synergiques ont été déterminés par une ANOVA à deux voies (valeur p <0,05) [20].
Test de consommation d’oxygène
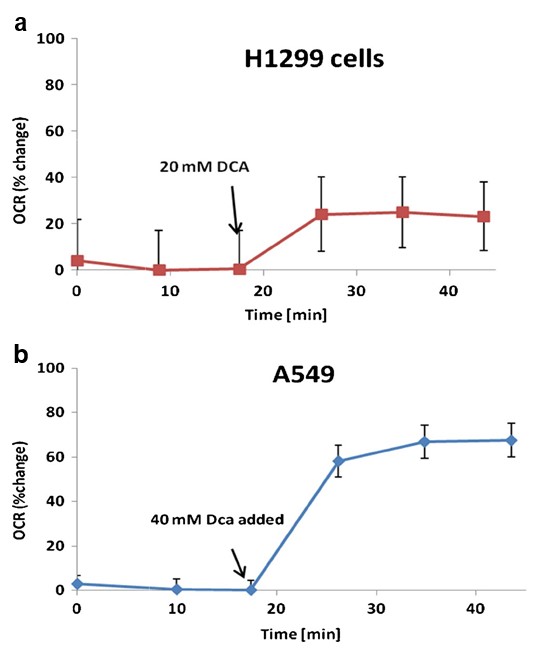
La consommation d’oxygène a été utilisée comme marqueur de substitution de l’activité mitochondriale. Les mesures ont été effectuées à l’aide d’un analyseur de flux extracellulaire XF24 (Seahorse Biosciences). Cet appareil utilise des capteurs optiques à fluorescence et des plaques multi-puits personnalisées pour effectuer des mesures répétées de la consommation d’oxygène de cellules intactes se développant en monocouche. Les cellules H1299 et A549 ont été ensemencées dans des plaques de culture cellulaire Seahorse XF24 à raison de 10 000 et 20 000 cellules par puits, respectivement, dans un milieu de croissance. Les cellules ont été incubées pendant 48 heures à 37 °C dans 5 % deCO2. Ensuite, les cellules ont été lavées et transférées dans un milieu de test XF/PBS, et incubées pendant 60 min à 37 °C sansCO2 avant de commencer l’expérience. Après avoir établi les taux de consommation d’oxygène de base, on a ajouté 20 et 40 mM de DCA aux cellules H1299 et A549, respectivement. Les mesures de la consommation d’oxygène ont été poursuivies pendant 25 minutes. Les données ont été acquises à partir d’au moins trois plaques répétées par lignée cellulaire. Les résultats sont représentés en pourcentage du taux de respiration de base.
Analyse statistique
La signification statistique a été évaluée à l’aide du test t de Student. La synergie a été évaluée par une ANOVA à deux voies [20]. Une valeur de p< 0,05 a été considérée comme significative.
Résultats
Le DCA radiosensibilise les cellules NSCLC
Le dichloroacétate(DCA) est connu pour augmenter l’activité mitochondriale [10,15] et il a été démontré précédemment qu’il agissait comme un radiosensibilisateur sur plusieurs lignées cellulaires cancéreuses dérivées de tumeurs de la prostate, colorectales et cérébrales [21, 22]. Nous avons étudié ici son effet sur deux lignées cellulaires dérivées du cancer du poumon non à petites cellules (CPNPC), à savoir A549 (adénocarcinome) et H1299 (carcinome à grandes cellules). Les concentrations de DCA utilisées pour chaque lignée cellulaire correspondaient aux valeurs respectives de la CI50 (A549 : 63 mM et H1299 : 36 mM ; Fig. 3a et b).
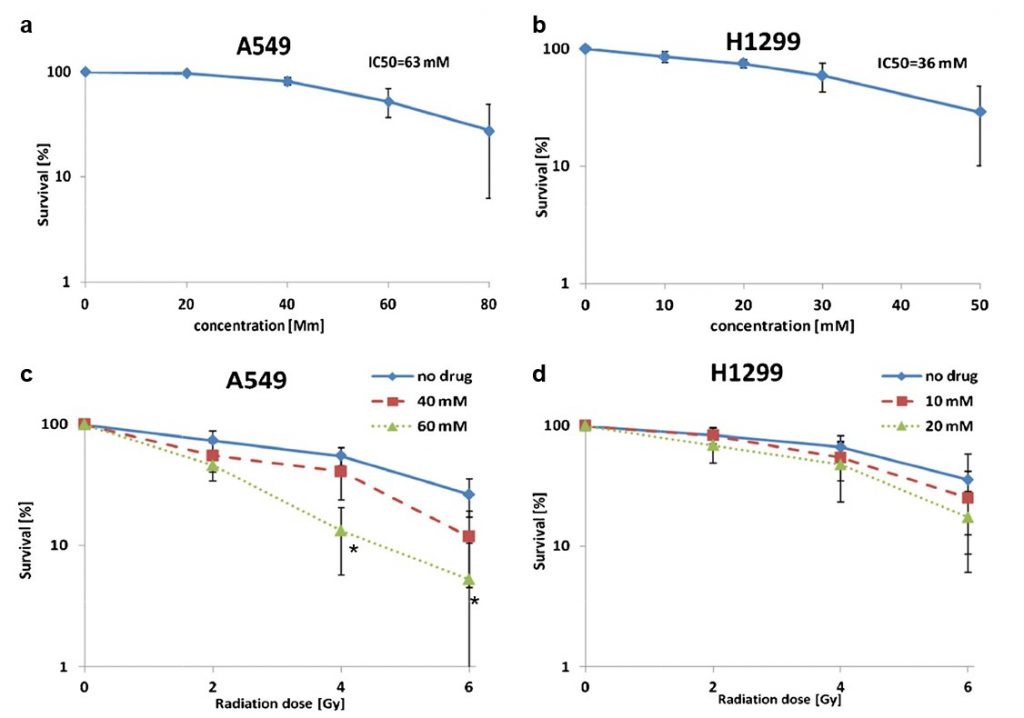
Nous avons constaté que l’exposition préalable au rayonnement avec le DCA augmentait la mort cellulaire dans les échantillons de cellules A549 et H1299. L’effet maximal dans les cellules A549 a été observé à 4 Gy, où le traitement au DCA (40 et 60 mM) a réduit la survie cellulaire de ~55 à 40 % (p= 0,27) et 13 % (p= 0,004), respectivement (Fig. 3c). Dans les cellules H1299, l’effet maximal a été atteint à 6 Gy. À cette dose de rayonnement, le traitement au DCA (10 et 20 mM) a réduit la survie des cellules de 35 % (rayonnement seul) à 25 et 17 %, respectivement (non significatif sur le plan statistique ; figure 3d). La pré-exposition des cellules A549 au DCA (60 mM) a multiplié par quatre l’effet cytotoxique du rayonnement de 4 Gy par rapport au rayonnement seul. Une analyse ANOVA à deux voies a indiqué un effet synergique uniquement dans les cellules A549.
Le DCA augmente la consommation d’oxygène dans les cellules A549 et H1299
Une augmentation du taux de consommation d’oxygène cellulaire (OCR) est une indication d’une activité mitochondriale accrue [23]. Nous avons constaté que le DCA augmente le TCO dans les cellules A549 et H1299 de 60 % (p= 0,0037) et de 20 % (p= 0,0039), respectivement, par rapport au TCO de base (figure 4).
Discussion
En testant la sensibilité aux rayonnements avec et sans pré-exposition au dichloroacétate (DCA), nous avons découvert que l’activation mitochondriale par le DCA peut radiosensibiliser deux lignées cellulaires distinctes dérivées du cancer du poumon non à petites cellules (CPNPC) (c’est-à-dire A549 et H1299). Dans cette étude de preuve de concept, une large gamme de concentrations de DCA a été utilisée, afin d’adapter la dose à la valeur de la CI50 de chaque cellule. L’effet synergique du DCA s’est avéré statistiquement significatif dans les cellules A549, alors qu’il n’était pas très significatif dans les cellules H1299. Il est possible qu’un effet encore plus marqué puisse être obtenu avec des doses plus élevées de DCA. Nous avons également montré que le DCA augmente la consommation d’oxygène dans les cellules A549 et H1299, ce qui est un marqueur de substitution pour une activité mitochondriale accrue.
Les rayonnements ionisants régulent à la hausse la fonction de la chaîne de transport d’électrons mitochondriale et augmentent le potentiel de la membrane mitochondriale et la production d’ATP [9]. L’hyperactivation mitochondriale par le DCA augmente encore la quantité et l’ampleur des radicaux libres (ROS) libérés par la mitochondrie elle-même [13]. Ces facteurs peuvent expliquer l’effet synergique observé par la pré-exposition des cellules au DCA avant le début de l’irradiation.
Atkinson et al. [15] ont montré que l’inhibition de la cytochrome c peroxydase et la libération de cytochrome c par les mitochondries atténuent la mort cellulaire induite par les rayonnements. Lee et al. [24] ont observé une diminution de l’inhibition de la mort cellulaire médiée par les ROS en bloquant la réponse anti-oxydante dépendante du facteur nucléaire érythroïde 2 (nrf2), ce qui augmente la radiosensibilité des cellules H1299, A549 et H640. Ces études ont montré la capacité d’affecter la réaction cellulaire aux rayonnements en interférant avec les processus liés au cytochrome c et aux ROS, ce qui renforce notre hypothèse selon laquelle le DCA radiosensibilise les cellules cancéreuses par une activation mitochondriale qui permet la libération de ROS.
Cao et al. [21] ont également signalé que le DCA agit comme un radiosensibilisateur dans les cellules cancéreuses de la prostate. Zwicker et al. [22] ont constaté que le DCA agit comme un radiosensibilisateur in vitro mais pas in vivo dans les cellules WIDR (colorectales) et LN18 (gliome). L’absence de synergie dans le modèle de xénogreffe WIDR in vivo a été expliquée par un effet tampon de l’environnement, qui a pu permettre aux cellules tumorales de maintenir leur programme métabolique glycolytique. Des études démontrant l’effet in vivo du DCA et des radiations dans le NSCLC restent encore à réaliser.
La radiothérapie est une thérapie courante pour les patients atteints de cancer du poumon. Les dommages considérables qu’elle cause aux tissus normaux environnants limitent toutefois son application. Au cours des deux dernières décennies, les inhibiteurs pharmacologiques des molécules de signalisation qui régulent le potentiel apoptotique se sont révélés prometteurs en tant que radiosensibilisateurs in vitro [25,29]. Bien que le DCA ait été signalé comme radiosensibilisateur dans le cancer de la prostate [21], son utilisation clinique dans le NSCLC n’a pas encore été prouvée. Nous montrons ici la capacité du DCA à diminuer les doses de radiation dans les cellules A549 et H1299 du NSCLC sans compromettre le niveau d’effet. En outre, en montrant que le traitement au DCA augmente la consommation d’oxygène des cellules A549 et H1299, nous avons mis en évidence le potentiel du DCA à agir comme un activateur mitochondrial. Par conséquent, nous concluons que l’activation mitochondriale peut jouer un rôle dans la thérapie du NSCLC lorsqu’elle est intégrée à la radiothérapie. En outre, nous émettons l’hypothèse que l’induction d’une activité mitochondriale accrue, c’est-à-dire la libération accrue de radicaux libres et l’apoptose par le DCA, est la principale cause du potentiel du DCA dans la radiosensibilisation des cellules NSCLC.
En résumé, nous montrons que l’induction mitochondriale in vitro par le DCA radiosensibilise de manière significative et synergique les cellules A549 NSCLC. Cette observation justifie une évaluation plus approfondie dans un contexte in vivo.
Remerciements
Les auteurs remercient le Dr Shoshana Paglin (Sheba Medical Center) pour ses conseils et ses discussions fructueuses concernant ce travail.
Ce travail a été réalisé dans le cadre de l’accomplissement partiel des exigences de la thèse de doctorat en médecine de la Faculté de médecine Sackler de l’Université de Tel Aviv et a été soutenu par le Centre médical Sheba. (M. Ronen Shavit)
Conflit d’intérêt
Les auteurs déclarent ne pas avoir de conflit d’intérêts.
RÉFÉRENCES
1 R. Siegel, C. DeSantis, K. Virgo, K. Stein, A. Mariotto, T. Smith, D. Cooper, T. Gansler, C. Lerro, S. Fedewa, C. Lin, C. Leach, R.S. Cannady, H. Cho, S. Scoppa, M. Hachey, R. Kirch, A. Jemal, E. Ward, Cancer treatment and survivorship statistics, 2012. CA Cancer J. Clin. 62(4), 220-241 (2012)2 Q. Wu, Y.F. Chen, J. Fu, Q.H. You, S.M. Wang, X. Huang, X.J. Feng, S.H. Zhang, Short hairpin RNA-mediated down-regulation of CENP-A attenuates the aggressive phenotype of lung adenocarcinoma cells. Cell. Oncol. 37(6), 399-407 (2014)
3 A. Koren, H. Motaln, T. Cufer, Les cellules souches du cancer du poumon : une perspective biologique et clinique. Cell. Oncol. 36(4), 265-275 (2013)
4 N. Peled, M.W. Wynes, N. Ikeda, T. Ohira, K. Yoshida, J. Qian, M. Ilouze, R. Brenner, Y. Kato, C. Mascaux, F.R. Hirsch, Insulin-like growth factor-1 receptor (IGF-1R) as a biomarker for resistance to the tyrosine kinase inhibitor gefitinib in non-small cell lung cancer. Cell. Oncol. 36(4), 277-288 (2013)
5 M.V. Graham, J.A. Purdy, B. Emami, W. Harms, W. Bosch, M.A. Lockett, C.A. Perez, Clinical dose-volume histogram analysis for pneumonitis after 3D treatment for non-small cell lung cancer (NSCLC). Int. J. Radiat. Oncol. Biol. Phys. 45(2), 323-329 (1999)
6 H. Vakifahmetoglu, M. Olsson, B. Zhivotovsky, Death through a tragedy : mitotic catastrophe. Cell Death Differ. 15(7), 1153-1162 (2008)
7 J. Thoms, R.G. Bristow, DNA repair targeting and radiotherapy : a focus on the therapeutic ratio. Semin. Radiat. Oncol. 20(4), 217-222 (2010)
8 C. Coleman, Beneficial liaisons : radiobiology meets cellular and molecular biology. Radiother Oncol : J. Eur. Soc. Ther. Radiol. Oncol. 28(1), 1-15 (1993)
1 T. Yamamori, H. Yasui, M. Yamazumi, Y. Wada, Y. Nakamura, H. Nakamura, O. Inanami, Ionizing radiation induces mitochondrial reactive oxygen species production accompanied by upregulation of mitochondrial electron transport chain function and mitochondrial content under control of the cell cycle checkpoint. Free Radic. Biol. Med. 53(2), 260-270 (2012)
10 I. Szumiel, Stress oxydatif, changements épigénétiques et instabilité génomique induits par les rayonnements ionisants : le rôle pivot des mitochondries. Int J Radiat Biol. 1-55 (2014)
11 B.A. Rupnow, S.J. Knox, The role of radiation-induced apoptosis as a determinant of tumor responses to radiation therapy. Apoptosis 4(2), 115-143 (1999)
12 R.A. Cairns, I.S. Harris, T.W. Mak, Regulation of cancer cell metabolism. Nat. Rev. Cancer 11(2), 85-95 (2011)
13 E.D. Michelakis, L. Webster, J.R. Mackey, Dichloroacetate (DCA) as a potential metabolic targeting therapy for cancer. Br. J. Cancer 99(7), 989-994 (2008)
14 X. Wang, S. Peralta, C.T. Moraes, Mitochondrial alterations during carcinogenesis : a review of metabolic transformation and targets for anticancer treatments. Adv. Cancer Res. 119, 127-160 (2013)
15 J. Atkinson, A.A. Kapralov, N. Yanamala, Y.Y. Tyurina, A.A. Amoscato, L. Pearce, J. Peterson, Z. Huang, J. Jiang, A.K. Samhan-Arias, A. Maeda, W. Feng, K. Wasserloos, N.A. Belikova, V.A. Tyurin, H. Wang, J. Fletcher, Y. Wang, I.I. Vlasova, J. Klein-Seetharaman, D.A. Stoyanovsky, H. Bayir, B.R. Pitt, M.W. Epperly, J.S. Greenberger, V.E. Kagan, A mitochondria-targeted inhibitor of cytochrome c peroxidase mitigates radiation-induced death. Nat. Commun. 2, 497 (2011)
16 S.H. Kim, Y.H. Yoo, J.H. Lee, J.W. Park, Mitochondrial NADP(+)-dependent isocitrate dehydrogenase knockdown inhibes tumorigenicity of melanoma cells. Biochem. Biophys. Res. Commun. 451(2), 246-251 (2014)
17 Z. Tatarkova, S. Kuka, M. Petras, P. Racay, J. Lehotsky, D. Dobrota, P. Kaplan, Why mitochondria are excellent targets for cancer therapy. Klin. Onkol. 25(6), 421-426 (2013)
18 S. Bonnet, S.L. Archer, J. Allalunis-Turner, A. Haromy, C. Beaulieu, R. Thompson, C.T. Lee, G.D. Lopaschuk, L. Puttagunta, G. Harry, K. Hashimoto, C.J. Porter, M.A. Andrade, B. Thebaud, E.D. Michelakis, A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11(1), 37-51 (2007)
19 N.A. Franken, H.M. Rodermond, J. Stap, J. Haveman, C. van Bree, Clonogenic assay of cells in vitro. Nat. Protoc. 1(5), 2315-2319 (2006)
20 B.K. Slinker, The statistics of synergism. J. Mol. Cell. Cardiol. 30(4), 723-731 (1998)
21 W. Cao, S. Yacoub, K.T. Shiverick, K. Namiki, Y. Sakai, S. Porvasnik, C. Urbanek, C.J. Rosser, Dichloroacetate (DCA) sensibilise in vitro les cellules cancéreuses de la prostate de type sauvage et sur-exprimant Bcl-2 aux radiations. Prostate 68(11), 1223-1231 (2008)
22 F. Zwicker, A. Kirsner, P. Peschke, F. Roeder, J. Debus, P.E. Huber, K.J. Weber, Dichloroacetate induit une radiosensibilité spécifique aux tumeurs in vitro mais atténue le retard de croissance tumorale induit par les radiations in vivo. Strahlenther. Onkol. 189(8), 684-692 (2013)
23I. Papandreou, R.A. Cairns, L. Fontana, A.L. Lim, N.C. Denko, HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 3(3), 187-197 (2006)
24 S. Lee, M.J. Lim, M.H. Kim, C.H. Yu, Y.S. Yun, J. Ahn, J.Y. Song, An effective strategy for increasing the radiosensitivity of Human lung Cancer cells by blocking Nrf2-dependent antioxidant responses. Free Radic. Biol. Med. 53(4), 807-816 (2012)
25 S.J. Chmura, H.J. Mauceri, S. Advani, R. Heimann, M.A. Beckett, E. Nodzenski, J. Quintans, D.W. Kufe, R.R. Weichselbaum, Decreasing the apoptotic threshold of tumor cells through protein kinase C inhibition and sphingomyelinase activation increases tumor killing by ionizing radiation. Cancer Res. 57(19), 4340-4347 (1997)
26 V. Bhardwaj, Y. Zhan, M.A. Cortez, K.K. Ang, D. Molkentine, A. Munshi, U. Raju, R. Komaki, J.V. Heymach, J. Welsh, L’inhibiteur de C-Met MK-8003 radiosensibilise les cellules cancéreuses pulmonaires non à petites cellules exprimant c-Met et radio-induites. J. Thorac. Oncol. 7(8), 1211-1217 (2012)
27 E.J. Bernhard, G. Kao, A.D. Cox, S.M. Sebti, A.D. Hamilton, R.J. Muschel, W.G. McKenna, The farnesyltransferase inhibitor FTI-277 radiosensitizes H-ras-transformed rat embryo fibroblasts. Cancer Res. 56(8), 1727-1730 (1996)
28 H.J. Boeckman, K.S. Trego, J.J. Turchi, Cisplatin sensitizes cancer cells to ionizing radiation via inhibition of nonhomologous end joining. Mol. Cancer Res. 3(5), 277-285 (2005)
29 N. Balaban, J. Moni, M. Shannon, L. Dang, E. Murphy, T. Goldkorn, The effect of ionizing radiation on signal transduction : antibodies to EGF receptor sensitize A431 cells to radiation. Biochim. Biophys. Acta 1314(1-2), 147-156 (1996)
Contenu connexe :