Allison B. Haugrud, Yongxian Zhuang, Joseph D. Coppock, W. Keith Miskimins
Cancer Biology Research Center, Sanford Research, 2301 E. 60th St North, Sioux Falls, SD 57104, USA
e-mail : [email protected]
Reçu : 24 avril 2014
Accepté : 5 septembre 2014
Publié : 12 septembre 2014
Résumé
Le métabolisme unique des cellules du cancer du sein offre un intérêt pour l’exploitation thérapeutique de ce phénomène. La metformine, un traitement prometteur du cancer du sein, cible le complexe I de la chaîne de transport d’électrons, conduisant à une accumulation d’espèces réactives de l’oxygène (ERO) qui finissent par entraîner la mort cellulaire. L’inhibition du complexe I entraîne la production de lactate, un sous-produit métabolique déjà fortement produit par les cellules cancéreuses reprogrammées et associé à un mauvais pronostic. Alors que la metformine reste un traitement anticancéreux prometteur, nous avons cherché un agent complémentaire pour augmenter les effets apoptotiques de la metformine tout en atténuant la production de lactate, ce qui pourrait améliorer considérablement l’efficacité. Le dichloroacétate (DCA) est un médicament bien établi utilisé dans le traitement de l’acidose lactique qui fonctionne par l’inhibition de la pyruvate déshydrogénase kinase (PDK) favorisant le métabolisme mitochondrial. Notre objectif était d’examiner la synergie et les mécanismes par lesquels ces deux médicaments tuent les cellules du cancer du sein. Des lignées cellulaires ont été soumises aux traitements indiqués et analysées pour la mort cellulaire et divers aspects du métabolisme. La mort cellulaire et la production de ROS ont été analysées à l’aide de la cytométrie en flux, de l’analyse Western blot et de méthodes de comptage des cellules. Des images de cellules ont été prises avec la microscopie à contraste de phase ou la microscopie confocale. Le métabolisme des cellules a été analysé à l’aide de l’analyseur Seahorse XF24, des dosages de lactate et de l’analyse du pH. Nous montrons que lorsque le DCA et la metformine sont utilisés en combinaison, une induction synergique de l’apoptose des cellules du cancer du sein se produit. Les dommages oxydatifs induits par la metformine sont renforcés par le DCA par le biais de l’inhibition de la PDK1 qui diminue également la production de lactate favorisée par la metformine. Nous démontrons que le DCA et la metformine se combinent pour induire de manière synergique une apoptose dépendante de la caspase impliquant des dommages oxydatifs avec une atténuation simultanée de la production de lactate favorisée par la metformine. Des combinaisons innovantes telles que la metformine et le DCA sont prometteuses pour l’élargissement des thérapies du cancer du sein.
Mots clés : Metformine ; Dichloroacétate ; Cancer du sein ; Lactate ; Apoptose
© Springer Science+Business Media New York 2014
INTRODUCTION
Le métabolisme du cancer est en train de devenir un domaine prometteur pour le développement de nouvelles approches thérapeutiques. Par rapport aux cellules normales dont elles dérivent, les cellules cancéreuses sont métaboliquement reprogrammées, utilisant préférentiellement la glycolyse même dans des conditions d’oxygène suffisant, un phénomène connu sous le nom d’effet Warburg [1]. Pour compenser la perte d’ATP résultant de la glycolyse préférentielle (par rapport à la progression par phosphorylation oxydative), les cellules cancéreuses régulent à la hausse les gènes codant pour les transporteurs de glucose et les enzymes glycolytiques comme la pyruvate déshydrogénase kinase (PDK) et la lactate déshydrogénase (LDH). Ce taux élevé d’absorption de glucose et la modification du métabolisme ne fournissent pas seulement de l’ATP et permettent aux cellules de survivre dans des conditions hypoxiques ; ils fournissent également des éléments de construction biosynthétiques tels que des intermédiaires et des substrats pour la production d’acides aminés, de NADPH et de ribose-5-phosphate, qui sont essentiels à la synthèse des nucléotides, des protéines et des membranes nécessaires aux cellules qui se divisent rapidement. Cela signifie également que le cycle TCA mitochondrial génère un pourcentage plus faible d’ATP, permettant au citrate d’être utilisé dans la biosynthèse des acides gras et des lipides pour la production de nouvelles membranes [2]. Une grande partie du lactate dérivé de la glycolyse est absorbée par les cellules environnantes avec un recyclage réciproque pour soutenir la croissance de la tumeur et la résistance aux mécanismes de mort cellulaire apoptotique [2, 3]. Le lactate produit par les tumeurs peut perturber le métabolisme des lymphocytes T et la présentation des antigènes par les cellules dendritiques, ce qui entraîne une évasion immunitaire pour la tumeur [4, 5]. Ainsi, les niveaux élevés d’utilisation du glucose et de glycolyse offrent aux cellules cancéreuses de nombreux avantages. Cependant, il pourrait également être possible d’exploiter le profil métabolique unique du cancer à des fins thérapeutiques. Dans cette étude, nous avons examiné l’activité et l’interaction de deux médicaments ciblant le métabolisme, la metformine et le dichloroacétate (DCA), afin de déterminer leurs effets sur la croissance et la survie des cellules du cancer du sein.
Le chlorhydrate de metformine (chlorhydrate de 1,1-diméthylbiguanide) est un médicament oral largement utilisé dans le traitement du diabète de type 2. Des études ont montré que les patients diabétiques sous metformine ont une incidence plus faible de cancer et de décès associés que les patients qui ne prennent pas de metformine. Dans l’une de ces études, des patientes diabétiques atteintes d’un cancer du sein et traitées à la metformine ont eu une réponse nettement meilleure à la chimiothérapie néoadjuvante que les patientes non traitées à la metformine [6-8]. Des études in vitro ont conclu que la metformine inhibe la croissance de nombreux types de cellules cancéreuses, notamment celles des cancers du sein, du côlon, de la prostate, de l’ovaire et des gliomes [9-12]. La metformine est connue pour activer la protéine kinase activée par l’AMP (AMPK), ce qui entraîne une inhibition de la synthèse des protéines et de la croissance cellulaire [13]. Cependant, l’activation de l’AMPK ne suffit pas à elle seule à entraîner la mort cellulaire apoptotique [14]. Des études ont montré que la metformine s’accumule dans les mitochondries et inhibe légèrement le complexe I de la chaîne de transport des électrons, un événement qui se produit en amont de l’activation de l’AMPK [15-18]. Lorsque le complexe I est inhibé, le passage entravé des électrons entraîne la production de superoxyde dans la matrice mitochondriale, ce qui endommage les protéines, les lipides et les acides nucléiques mitochondriaux. Dans les études où il a été démontré que la metformine favorise la mort cellulaire, l’apoptose est la principale voie d’action [10, 12, 19]. Nous avons précédemment montré que la metformine induit une mort cellulaire dépendante de la caspase et de la poly(ADP-ribose) polymérase (PARP) dans la plupart des lignées cellulaires de cancer du sein, tout en étant non cytotoxique pour les cellules épithéliales mammaires non transformées [20]. La mort cellulaire dépendante de la poly(ADP-ribose) polymérase était associée à des altérations majeures de la forme et de la fonction des mitochondries, ce qui a permis de conclure que les lésions mitochondriales dans les cellules cancéreuses sont un médiateur clé de la mort cellulaire induite par la metformine. Sur la base de ces observations, nous avons émis l’hypothèse que les composés qui favorisent le métabolisme oxydatif mitochondrial renforceraient les dommages mitochondriaux induits par la metformine et agiraient en synergie avec la metformine pour tuer les cellules cancéreuses. Comme le traitement par la metformine favorise également la production de lactate [21], l’idéal serait qu’un tel composé combatte également cet effet.
Le dichloroacétate est également un médicament disponible par voie orale dont la pharmacocinétique a été bien étudiée et qui a été testé pour le traitement de l’acidose lactique (un effet secondaire potentiel de la metformine) et des déficiences mitochondriales [27]. Le dichloroacétate est un inhibiteur de la PDK qui phosphoryle la pyruvate déshydrogénase (PDH), la rendant inactive [23]. La pyruvate déshydrogénase est l’enzyme responsable de la catalyse de la transformation du pyruvate en acétyl-CoA pour son entrée dans le cycle mitochondrial de l’acide tricarboxylique (TCA) et la phosphorylation oxydative. Dans les cellules cancéreuses, l’activité de la PDK est souvent élevée, agissant comme un garde-barrière pour réduire le flux de pyruvate du cytoplasme vers le métabolisme mitochondrial. On pense qu’il s’agit d’un élément important de la reprogrammation métabolique des cellules cancéreuses, qui entraîne une réduction de l’oxydation du glucose et de la production de lactate [24-26]. En inhibant la PDK, le DCA augmente l’activité de la PDH, permettant au pyruvate d’entrer dans le cycle TCA plutôt que d’être converti en lactate et sécrété [27].
Dans cette étude, nous avons examiné l’activité antitumorale et l’interaction de deux médicaments ciblant le métabolisme, la metformine et le DCA. Nous montrons que le DCA renforce la cytotoxicité de la metformine sur les cellules cancéreuses du sein par un mécanisme impliquant des dommages oxydatifs, tout en réduisant simultanément la production de lactate par la metformine, ce qui peut présenter un double avantage thérapeutique.
Méthodes
Produits chimiques et réactifs
Les produits chimiques, réactifs et kits suivants ont été achetés chez Sigma-Aldrich, sauf indication contraire : metformine (1,1-diméthylbiguanide), dichloroacétate de sodium, solution de bleu trypan 0.solution de bleu trypan à 4 %, milieu de montage Vectashield pour fluorescence contenant du 4,6 diamidino-2-phénylindole (DAPI) (Vector Laboratories), kit de dosage du lactate (Eton Biosciences), inhibiteur de caspase OPH-109 (MP Biomedicals), bleu brillant de Coomassie R250 (Bio-Rad Laboratories), paraformaldéhyde, SYTOX® Green (Life Technologies), Triton X-100 (Eastman), et inhibiteur de PARP II INH2BP (Epigentek).
Culture cellulaire
Les lignées cellulaires de cancer du sein humainMCF7et T47D et les cellules épithéliales mammaires humaines MCF10A ont été achetées auprès de l’ATCC. La lignée cellulaire 66CL4 de carcinome mammaire de souris a été fournie par le Dr Fred Miller (Karmanos Cancer Institute, Detroit, MI). Dès réception des lignées cellulaires, les cellules ont été immédiatement mises en culture et expansées pour préparer des stocks d’ampoules congelées. Les cellules ont été passées pendant 2 à 3 mois au maximum avant d’établir de nouvelles cultures à partir des ampoules congelées de passage précoce. Les lignées cellulaires ont fait l’objet d’un contrôle régulier de la contamination par les mycoplasmes et ont été vérifiées comme étant exemptes de mycoplasmes par IDEXX RADIL Laboratories. Les cellules ont été maintenues dans du milieu Dulbecco’s modified Eagle’s (DMEM) avec 10 % de sérum bovin fœtal, 100 U/ml de pénicilline et 100 µg/ml de streptomycine. Les cellules ont été incubées dans un incubateurCO2 humidifié à 37 °C.
Test d’exclusion au bleu Trypan
Les lignées cellulairesMCF7et T47D ont été placées sur des boîtes de 35 mm. Les cellules ont été traitées avec la metformine et le DCA aux concentrations indiquées ou avec le véhicule. Après la période de temps indiquée, le milieu a été collecté pour conserver les cellules mortes flottantes. Les cellules attachées ont été lavées avec une solution saline tamponnée au phosphate (PBS), qui a été regroupée avec le milieu collecté. Les cellules ont ensuite été récoltées par trypsinisation et ajoutées au pool. Une solution de bleu trypan a ensuite été utilisée pour colorer les cellules mortes dans une proportion de 1:1, et les cellules ont été comptées au moyen d’un hémacytomètre.
Western blotting
Les cellules ont été placées sur des boîtes de 35 mm. Après le traitement pendant la période indiquée, les cellules ont été récoltées dans le milieu pour recueillir les cellules vivantes et mortes. Les cellules ont été pelletées, lavées avec du PBS et re-pelletées. Le culot cellulaire a ensuite été lysé par l’ajout de 1× tampon d’échantillon de dodécylsulfate de sodium (SDS) [2,5 mM Tris-HCl (pH 6,8), 2,5 % SDS, 100 mM dithiothréitol, 10 % glycérol, 0,025 % bleu de bromophénol]. Des quantités égales de protéines ont été séparées sur un gel de SDS-polyacrylamide à 8,5 %. Les protéines ont été transférées sur des membranes Immobilon P (Millipore) avec un appareil Bio-Rad Trans-blot en utilisant un tampon de transfert [48 mM Tris-HCl, 39 mM glycine]. Les membranes ont été immergées dans du lait sec non gras à 5 % dans une solution saline tamponnée au tris contenant du Tween 20 (TBS-T) [10 mM Tris-HCl (pH 7,5), 150 mM NaCl, 0,1 % Tween-20] avec l’anticorps indiqué pendant 3 h à température ambiante ou 4 °C pendant la nuit. Après un lavage soigneux avec du TBS-T, un anticorps secondaire approprié conjugué à la peroxydase de raifort (HRP) a été appliqué. La membrane a été lavée à nouveau, puis les protéines ont été détectées à l’aide du substrat chimiluminescent Super Signal West Pico (Pierce Biochemical). Pour l’isolement des mitochondries, (Fig. 3d) les cellules MCF7 ont été cultivées jusqu’à 95 % de confluence sur des plats de 150 mm. Un kit d’isolation des mitochondries pour cellules en culture (Pierce) a été utilisé pour isoler les mitochondries conformément au protocole du fabricant. Tous les blots ont été imagés à l’aide d’un système d’imagerie UVP. Les anticorps suivants ont été utilisés : Pyruvate déshydrogénase kinase1 (Abcam), PARP (Cell Signaling), anti-4-hydroxynonénal (Millipore), sous-unité du complexe I NDUFB8 (Mitosciences), GAPDH (Ambion), PDH (Abcam) et Phospho-PDHE1 alpha (Calbiochem).
Microscopie confocale
Les cellules ont été placées sur des lamelles de verre dans des boîtes de 35 mm et traitées comme indiqué. Les cellules ont été lavées dans du PBS, fixées avec du paraformaldéhyde à 4 % pendant 10 minutes, montées dans du milieu Vectashield avec du DAPI sur des lames de microscope standard, puis observées sur un microscope confocal Olympus FV1000 à 100×. L’établissement de lignées cellulaires MCF7 exprimant de manière stable pAcGFP1-Mito (Clontech ; un plasmide codant pour des protéines fluorescentes ciblées sur les mitochondries) a été décrit précédemment [20].
Dosage du lactate et analyse du pH
Les cellulesMCF7et 66CL4 placées dans des boîtes de 35 mm ont été cultivées jusqu’à 80 % de confluence et traitées comme indiqué dans des milieux sans rouge de phénol et sans carbonate. Les cellules ont été traitées pendant 4 à 6 heures. Le milieu des boîtes a été recueilli et la concentration de lactate mesurée à l’aide d’un kit de dosage du L-Lactate disponible dans le commerce (Eton Bioscience). Pour la lignée cellulaire 66Cl4, les cellules ont été comptées par hémacytomètre pour normaliser les niveaux de lactate. Un pH-mètre a été utilisé pour mesurer le pH du milieu.
Test de formation de colonies
Les cellulesMCF7ont été placées à raison de 500 cellules par boîte de 60 mm. Le jour suivant, les cellules ont été traitées comme indiqué. Après 12 jours d’incubation, les cellules ont été lavées avec du PBS et fixées avec de l’éthanol à 70 % pendant 5 minutes. Les colonies ont ensuite été colorées avec du bleu de Coomassie [40 % de méthanol, 12 % d’acide acétique glacial, 0,24 % de bleu de Coomassie], lavées, imagées et quantifiées à l’aide d’un système AlphaImager et du logiciel d’analyse d’image correspondant (AlphaInnotech, Santa Clara, CA).
Cytométrie en flux
Les cellulesMCF7ont été traitées comme indiqué. Les cellules ont ensuite été incubées avec MitoSOX (5 µM) pendant 15 min, lavées, trypsinisées, et 1 ml de DMEM à 10 % de FBS a été ajouté aux cellules. La détection des niveaux de MitoSOX dans un nombre égal de cellules par condition de traitement a été déterminée par cytométrie de flux en utilisant un cytomètre Accuri C6.
Inhibition de PDK1 à l’aide d’un siRNA
On-TARGET Plus Smartpool siRNA ciblant PDK1 et siRNA non ciblés ont été achetés auprès de Thermo Scientific. les cellules 66CL4 (250 000/plateau) ont été placées sur des plats de 35 mm. Le jour suivant, les cellules ont été transfectées avec les siRNA en utilisant Dharmafect (Thermo Scientific) selon le protocole du fabricant. La transfection a été répétée le jour suivant. Le jour suivant, les cellules ont été lavées et traitées comme indiqué dans du DMEM sans carbonate avec 10 % de FBS. Après 4 h, les cellules ont été comptées à l’aide de l’hémacytomètre, et les milieux ont été collectés pour mesurer le pH et les niveaux de lactate. Les cellules ont été collectées pour l’analyse des niveaux de PDK1 par western blotting.
Test des substances réactives de l’acide thiobarbiturique (TBARS)
Les cellulesMCF7ont été cultivées jusqu’à 100 % de confluence sur des boîtes de 150 mm. Les cellules ont été traitées comme indiqué pendant 24 h avec de la metformine (8 mM) ou du DCA (5 mM). Les cellules ont été récoltées dans du PBS, soniquées et les valeurs protéiques relatives ont été déterminées à l’aide du test Bradford. Les lysats ont été traités à l’aide du kit de dosage Quantichrom™ TBARS et lus sur un lecteur de plaques SpectraMax M5 (λ ex/em = 560 nm/585 nm).
Essai SYTOX® green
Les cellulesMCF7et T47D ont été placées sur des plaques à 96 puits et cultivées jusqu’à 90 % de confluence. Les cellules ont été traitées comme indiqué, suivies de l’ajout du colorant d’acide nucléique SYTOX® Green (10 µM) et d’une incubation de 20 min avant lecture sur un lecteur de plaques à fluorescence à un λ ex/em = 485/535 nm avec une coupure à 515 nm. Les cellules ont ensuite été perméabilisées avec du Triton X-100 (0,4 %) pendant 30 minutes, et une deuxième lecture a été effectuée pour déterminer le niveau total de coloration de l’ADN, un substitut du nombre total de cellules. Les valeurs de l’indice de combinaison ont été déterminées à l’aide du logiciel CalcuSyn.
Taux de consommation d’oxygène
Le taux de consommation d’oxygène (OCR) a été mesuré à l’aide d’un analyseur Seahorse XF24 conformément aux instructions du fabricant (Seahorse Bioscience, North Billerica, MA, USA). Les cellules T47D ont été placées à 40 000 cellules/puits dans des plaques à puits XF24. Le lendemain, les cellules ont été lavées et équilibrées avec un milieu sans tampon pendant 30 minutes à 37 °C dans un incubateursans CO2 avant d’être transférées dans l’analyseur XF24. Une mesure initiale de l’OCR a été obtenue, suivie de l’ajout de DCA via des ports d’injection dans les puits jusqu’à une concentration finale de 5 mM. L’OCR a été mesuré toutes les 30 min.
Analyse statistique
La comparaison de deux groupes a été effectuée à l’aide d’un test t non apparié avec correction de Welch généré dans le logiciel Graph Pad Prism (La Jolla, CA). Les valeurs P inférieures ou égales à 0,05 ont été considérées comme significatives.
Résultats
LeDCA et la metformine induisent de manière synergique l’apoptose des cellules du cancer du sein
Nous avons précédemment montré que la metformine induit deux types distincts de mort cellulaire après 3 jours de traitement dans la plupart des lignées cellulaires du cancer du sein, tout en étant non cytotoxique pour les cellules épithéliales mammaires normales [20]. L’induction de la mort cellulaire dans les cellules de cancer du sein était étroitement associée à l’altération de la structure mitochondriale, ce qui suggère que la metformine, un inhibiteur connu du complexe I de la chaîne de transport d’électrons mitochondriale, tue les cellules cancéreuses en altérant la fonction mitochondriale. Nous avons donc émis l’hypothèse que l’augmentation du transport d’électrons mitochondrial renforcerait la mort des cellules cancéreuses induite par la metformine. Comme le DCA favorise l’oxydation mitochondriale du pyruvate, nous avons d’abord examiné si le DCA pouvait renforcer la cytotoxicité induite par la metformine dans les lignées cellulaires du cancer du sein.
Les cellules de cancer du sein MCF7 et T47D ont été traitées avec de la metformine, du DCA ou leur combinaison. La concentration de metformine utilisée était de 8 mM, ce qui représente une dose de metformine physiologiquement pertinente, comme le montrent les travaux d’Owen et al [17]. Comme pour le DCA, de faibles concentrations millimolaires peuvent être atteintes dans le sérum des patients, la plage de 0,5 à 5 mM est donc cliniquement pertinente [27-29] et a été utilisée dans ces expériences. Le nombre de cellules vivantes et mortes a été déterminé par le test d’exclusion au bleu trypan (Fig. 1a). La mort cellulaire a été mesurée dans les cellules MCF7 (Fig. 1a gauche) traitées pendant 2 jours, et les cellules T47D (Fig. 1adroite) ont été traitées pendant 4 jours. Dans les deux lignées cellulaires, la metformine a induit la mort cellulaire comme cela a été observé dans des expériences précédentes [20]. Cependant, la mort cellulaire était significativement augmentée par le traitement simultané par le DCA et la metformine, alors que le DCA seul avait des effets cytotoxiques minimes (Fig. 1a). Lorsque les cellules épithéliales mammaires non transformées MCF10A ont été soumises aux mêmes traitements, la mort cellulaire n’a pas été observée, comme le montre la figure 1b. Pour approfondir cette observation de la mort cellulaire dans les cellules cancéreuses, des titrages de dose et des tests de cytotoxicité SYTOX Green ont été réalisés (Fig. 1c). La synergie de l’association médicamenteuse a été testée par la méthode de Chou [30] à l’aide du logiciel CalcuSYN qui génère un indice de combinaison (IC) des médicaments, à partir duquel une synergie est indiquée si l’IC est inférieur à 1. Les cellules MCF7 (Fig. 1c gauche) avaient un IC de 0,00047 lorsqu’elles étaient traitées pendant deux jours par l’association de DCA et de metformine. Les cellules T47D avaient un IC de 9,762e-006 après 4 jours de traitement (Fig. 1c droite). Ces valeurs indiquent une forte cytotoxicité synergique avec la combinaison du DCA et de la metformine dans ces lignées cellulaires de cancer du sein.
Ensuite, les effets cytotoxiques observés du DCA (2,5 mM) et de la metformine (1 mM) ont été validés par des essais de formation de colonies. À ces concentrations, les deux médicaments seuls ont réduit la taille des colonies mais n’ont pas réduit de façon significative leur nombre (Fig. 1d, e). Cependant, une diminution significative du nombre de colonies a été observée lorsque les deux médicaments ont été combinés (Fig. 1e). Ces observations démontrent que le DCA augmente la cytotoxicité induite par la metformine dans les cellules cancéreuses du sein par rapport à l’un ou l’autre des médicaments seuls.
LeDCA favorise la phosphorylation oxydative par l’inhibition de PDK1 et atténue la production de lactate induite par la metformine
Pour confirmer que les effets observés du DCA sont dus à l’inhibition de la cible attendue, PDK1, nous montrons que le DCA diminue effectivement les niveaux de PDH phosphorylée par rapport au contrôle dans les cellules MCF7 et MCF10A par western blot (Fig. 2a). La stimulation de la pyruvate déshydrogénase à la suite d’un traitement au DCA devrait améliorer le métabolisme oxydatif du pyruvate. Cet effet a été étudié à l’aide d’un analyseur Seahorse XF24. Les ROC des cellules MCF7 témoins et des cellules traitées au DCA ont été mesurés (Fig. 2b). Les cellules traitées au dichloroacétate présentaient un ROC significativement plus élevé par rapport aux cellules témoins.
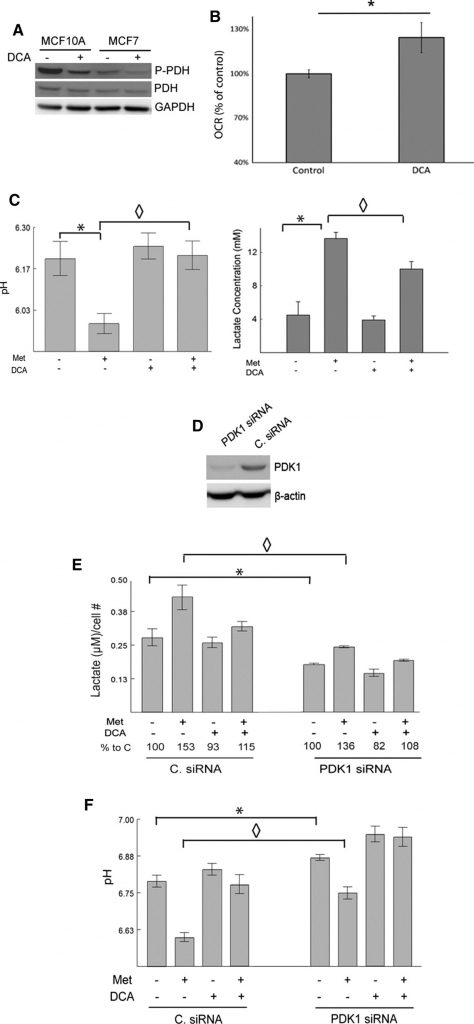
La metformine, en tant qu’inhibiteur léger du complexe I et amplificateur de l’activité AMPK, devrait stimuler les taux glycolytiques et augmenter la production de lactate [11,14,15,17]. En revanche, le DCA devrait rediriger le pyruvate vers le métabolisme mitochondrial au détriment de la production de lactate. Pour examiner cette hypothèse, le pH et les niveaux de lactate dans le milieu de culture des cellules MCF7 ont été mesurés après traitement par la metformine, le DCA ou les deux. Les cellules traitées à la metformine ont produit des niveaux de lactate significativement plus élevés que les contrôles non traités. Le dichloroacétate seul a eu de faibles effets mais a diminué de manière significative la production de lactate induite par la metformine. Le pH du milieu correspondait aux changements observés dans les niveaux de lactate (Fig. 2c). C’est-à-dire que la metformine a réduit de façon significative le pH du milieu et que cet effet a été largement éliminé par le DCA.
Pour étayer davantage le rôle de la PDK dans la production de lactate et l’inhibition de l’entrée du pyruvate dans le cycle TCA, la PDK a été désactivée à l’aide d’un siRNA dans les cellules 66CL4. Cette lignée cellulaire est dérivée d’un carcinome mammaire de souris et pourrait être utile pour transposer ces expériences dans un cadre préclinique lors de futures études. Les cellules ont été transfectées avec un siRNA qui cible PDK1 (PDK1siRNA) ou un siRNA de contrôle non ciblé (C. siRNA). Les niveaux de PDK1 ont été détectés par western blot, et les cellules transfectées par le siRNA PDK1 présentaient des niveaux de PDK1 nettement inférieurs à ceux des cellules transfectées par le siRNA C. (Fig. 2d). La répression de PDK1 par le siRNA a réduit la production de lactate et augmenté le pH du milieu, conformément aux fonctions connues de PDK1 (Fig. 2e, f). Le knockdown de PDK1 a diminué efficacement les changements induits par la metformine dans le lactate et le pH du milieu. Le dichloroacétate a encore diminué le lactate et augmenté le pH dans les cellules transfectées par le siRNA de PDK1. La capacité du DCA à inverser les effets de la metformine sur le lactate et le pH a été quelque peu atténuée lorsque PDK1 a été désactivé, ce qui suggère à nouveau que le DCA exerce ses effets par l’intermédiaire de PDK1. En résumé, on peut conclure que la perte de PDK1 par transfection par siRNA ou par inhibition par le DCA empêche la production de lactate induite par la metformine.
LeDCA associé à la metformine augmente les dommages oxydatifs et la mort cellulaire ultérieure dépendante de la caspase
La metformine inhibe le complexe I de la chaîne de transport des électrons, ce qui entrave le flux d’électrons et conduit à la production de superoxyde dans la matrice mitochondriale. Comme le DCA favorise l’entrée du pyruvate dans le cycle TCA, nous avons émis l’hypothèse que le DCA pourrait augmenter la production de ROS comme mécanisme potentiel de sa synergie cytotoxique avec la metformine. Le MitoSOX, un indicateur spécifique de la production de superoxyde mitochondrial, a coloré les cellules qui ont été analysées par cytométrie en flux. Après 1 h de traitement, ni la metformine (ligne rouge Fig. 3a, panneau de gauche) ni le DCA (non montré) n’ont augmenté la production de superoxyde mitochondrial. Cependant, le traitement combiné avec le DCA et la metformine a produit un déplacement vers la droite de l’intensité de la fluorescence, indiquant une production accrue de superoxyde. Après 3 h de traitement, les cellules traitées par la metformine ont commencé à produire des niveaux accrus de superoxyde mitochondrial (ligne rouge, figure 3a, panneau de droite) par rapport aux cellules non traitées (ligne noire). La combinaison du DCA et de la metformine a montré des niveaux encore plus élevés de coloration des superoxydes. Ces résultats indiquent que la metformine interfère avec le transport des électrons dans le complexe I, ce qui entraîne une production accrue de superoxyde, et que le DCA potentialise l’effet de la metformine.
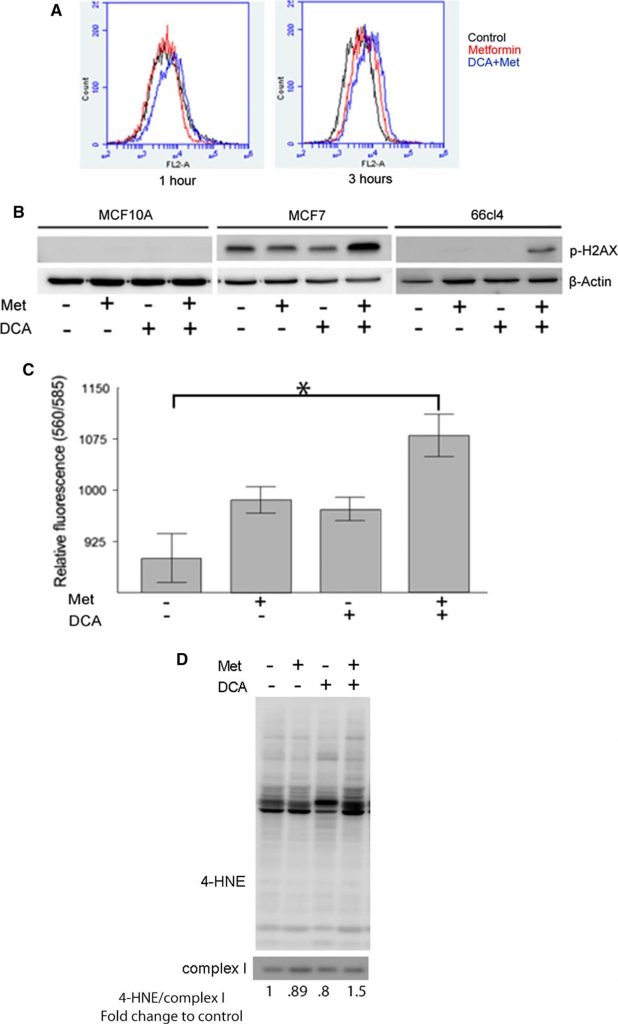
L’accumulation de superoxyde et d’autres ROS peut endommager les composants cellulaires, notamment les protéines, les membranes et l’ADN. Les lésions oxydatives de l’ADN peuvent entraîner des cassures double brin et la phosphorylation de l’histone H2AX (p-H2AX), qui est un marqueur de substitution utile de ces lésions de l’ADN [31]. Par conséquent, pour estimer les dommages à l’ADN associés à la production de ROS en réponse au traitement par la metformine et le DCA, les niveaux de H2AX phosphorylé peuvent être analysés. Les cellules épithéliales mammaires humaines MCF10A et les lignées cellulaires de cancer du sein MCF7 et 66CL4 ont été analysées par western blotting après traitement par DCA, metformine ou leur combinaison pendant 24 h (Fig. 3b). Dans les cellules MCF10A, aucune quantité détectable de p-H2AX n’a été observée, quel que soit le traitement. Dans les cellules MCF7, p-H2AX était présent dans les cellules non traitées et était considérablement augmenté par la combinaison de la metformine et du DCA, mais pas par l’un ou l’autre des médicaments seuls. Dans les cellules 66CL4, p-H2AX n’a été observé que dans les cellules co-traitées par le DCA et la metformine. Ces données suggèrent que le DCA et la metformine se combinent pour augmenter les dommages oxydatifs à l’ADN.
Comme deuxième niveau de preuve, nous avons mesuré la peroxydation lipidique qui se produit lorsque les radicaux libres endommagent les lipides des membranes cellulaires. Deux méthodes distinctes ont été utilisées pour estimer l’ampleur de l’oxydation des lipides après un traitement au DCA et à la metformine. Tout d’abord, le test TBARS qui mesure les sous-produits de la peroxydation lipidique et est utilisé pour évaluer l’étendue des dommages oxydatifs des membranes cellulaires causés par les ROS. Les TBARS ont augmenté dans les cellules MCF7 traitées soit par la metformine, soit par le DCA seul, et la plus forte augmentation a été observée lorsque les deux médicaments ont été combinés (Fig. 3c). La deuxième méthode utilisée pour estimer l’oxydation des lipides était un western blot des adduits protéiques 4-hydroxynonenol (4-HNE) qui sont dérivés de l’oxydation des acides gras insaturés dans les membranes cellulaires et forment des adduits covalents avec les protéines, fournissant une indication des dommages oxydatifs aux membranes cellulaires. Les extraits mitochondriaux de MCF7 provenant de cellules traitées avec du DCA, de la metformine ou les deux pendant 24 h ont été analysés pour déterminer les niveaux d’adduits protéiques 4-HNE (Fig. 3d). Une augmentation des niveaux d’adduits 4-HNE mitochondriaux a été observée uniquement dans les cellules traitées par la combinaison de metformine et de DCA. Cela confirme les résultats précédents selon lesquels l’association de ces médicaments entraîne une production accrue de ROS.
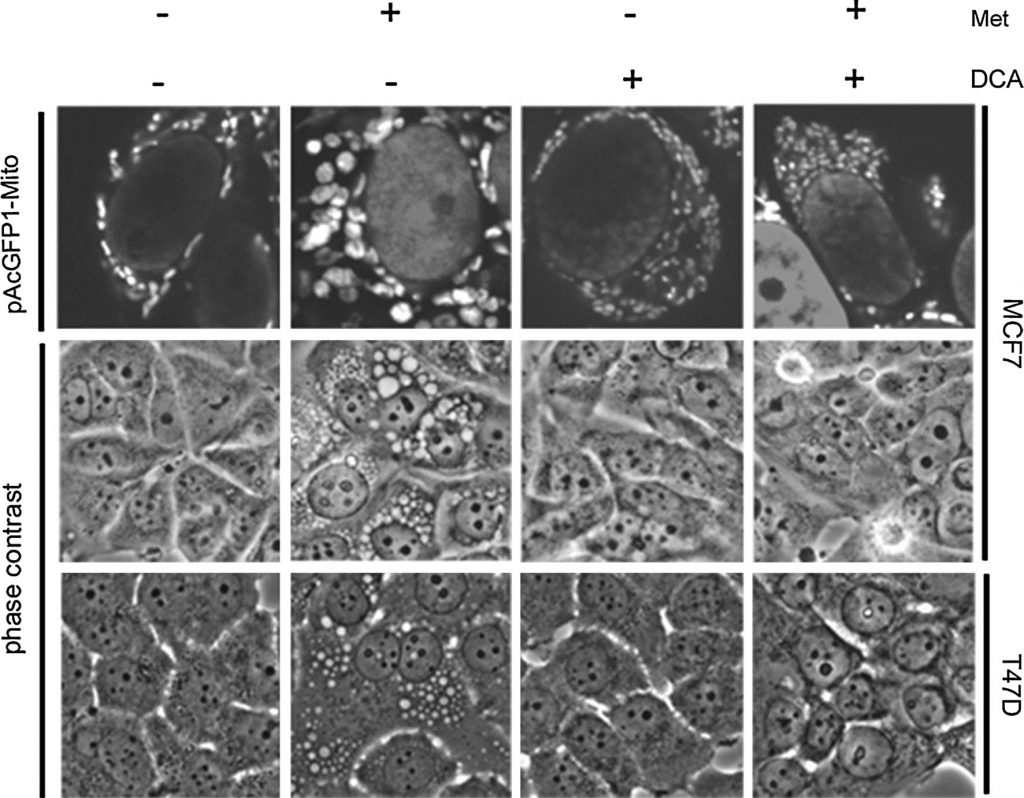
Nos données antérieures montrent que les cellules sensibles à la metformine développent de grandes vacuoles qui ont été déterminées par l’imagerie des mitochondries liées à la GFP et par la microscopie électronique comme étant des mitochondries structurellement altérées. Ces mitochondries altérées semblent être spécifiquement liées à une voie de mort cellulaire dépendant de la PARP qui est retardée dans le temps par rapport à la mort cellulaire apoptotique [20]. Comme le DCA agit en synergie avec la metformine dans la mort cellulaire, les effets du DCA sur la structure mitochondriale ont été étudiés. Une lignée cellulaire MCF7 stable exprimant une protéine fluorescente verte ciblée sur les mitochondries (pAcGFP1-Mito) a été développée. Les cellules ont été traitées pendant un jour avant d’être imagées par contraste de phase et microscopie confocale (Fig. 4). Dans les cellules traitées par la metformine seule, de grandes mitochondries ont été observées, comme cela avait été le cas précédemment [20]. En revanche, les cellules traitées au DCA semblaient avoir des mitochondries plus petites et plus nombreuses. Le dichloroacétate a également empêché l’élargissement des mitochondries provoqué par la metformine. Par contraste de phase, les mitochondries agrandies étaient apparentes dans les cellules traitées à la metformine alors qu’elles étaient absentes dans le traitement combiné. Une deuxième lignée cellulaire sensible à la metformine, T47D, a également été examinée. Là encore, des mitochondries hypertrophiées ont été observées lors du traitement à la metformine, alors qu’elles étaient absentes dans le traitement combiné.
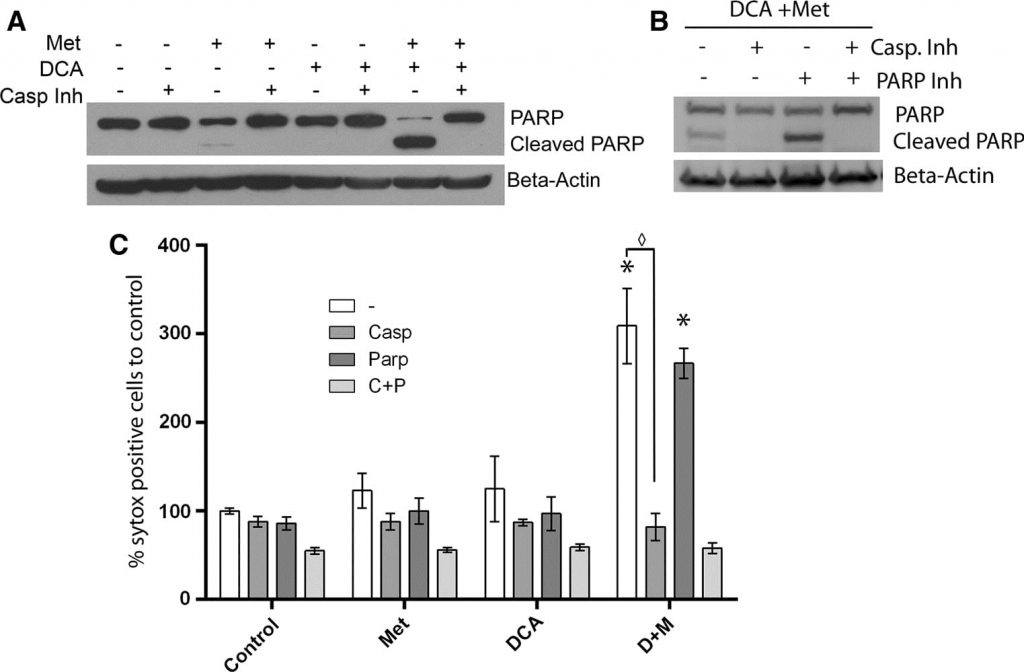
Dans nos travaux précédents, nous avons constaté que l’élargissement des mitochondries était corrélé à la mort cellulaire dépendante de la PARP, qui était retardée par rapport à la mort cellulaire apoptotique. Cela nous a incités à étudier les spécificités du mécanisme de mort cellulaire induit par le DCA combiné à la metformine [20], étant donné qu’aucune hypertrophie mitochondriale n’est observée. Pour caractériser la mort cellulaire induite dans les cellules co-traitées par la metformine et le DCA, les cellules MCF7 ont été traitées avec un inhibiteur de caspase irréversible à large spectre (Q-Val-Asp-OPh) en même temps que le DCA, la metformine ou les deux pendant une journée. Le Western blotting (Fig. 5a) a révélé une forte augmentation de la PARP clivée et une diminution de la PARP intacte dans les cellules co-traitées par le DCA et la metformine. L’ajout de l’inhibiteur de caspase a complètement bloqué le clivage de la PARP. Un effet similaire a été observé précédemment dans des cellules traitées par la metformine seule pendant 2,5 jours [20]. Cependant, au bout d’un jour, la metformine seule n’induit qu’une légère augmentation de la PARP clivée. Ces données indiquent donc que l’apoptose dépendante des caspases est accélérée par l’ajout de DCA à la metformine dans ces cellules de cancer du sein. Pour vérifier davantage si une mort dépendante de la PARP se produit lors du co-traitement du DCA et de la metformine, un inhibiteur de la PARP a également été utilisé. Un test SYTOX Green a été réalisé (figure 5c) deux jours après le traitement pour quantifier la mort cellulaire afin de révéler si l’inhibiteur PARP avait un effet additif à l’inhibiteur de caspase. L’inhibiteur PARP n’a eu aucun effet protecteur seul ni aucun effet additif avec l’inhibiteur de caspase. Un western blot a confirmé cette observation (figure 5b) et a révélé que l’inhibiteur de PARP n’a pas empêché le clivage de PARP. Comme l’élargissement des mitochondries est associé à la mort cellulaire dépendant de la PARP dans les cellules traitées par la metformine, l’absence d’élargissement des mitochondries lors de l’ajout de DCA suggère que l’apoptose se produit exclusivement de manière dépendante des caspases.
Discussion
La metformine est un médicament utilisé depuis longtemps dans le traitement du diabète de type 2. De même, le DCA a été étudié dans le cadre d’essais cliniques pour le traitement de l’acidose lactique et des déficiences mitochondriales. Il a déjà été démontré que la metformine ralentit la prolifération et favorise la mort cellulaire de nombreuses lignées de cellules cancéreuses par apoptose [9, 10, 12, 32]. La metformine se concentre dans les mitochondries où elle inhibe le complexe I de la chaîne de transport des électrons, provoquant un flux anormal d’électrons vers l’oxygène, produisant du superoxyde dans la matrice mitochondriale [22, Fig. 3]. La production d’espèces réactives de l’oxygène est problématique pour les cellules cancéreuses du sein car beaucoup d’entre elles ont une expression plus faible de la manganèse superoxyde dismutase (MnSOD) qui élimine normalement le superoxyde mitochondrial [33]. Bien que les effets pro-apoptotiques et inhibiteurs de croissance de la metformine sur le cancer soient remarquables, notre observation selon laquelle la metformine augmente la glycolyse, comme en témoigne la production accrue de lactate in vitro [21], peut limiter son efficacité en tant qu’agent solo. Comme des niveaux élevés de lactate ont été décrits comme favorisant les tumeurs et sont associés à de mauvais résultats cliniques [26, 34-38], nous avons cherché à atténuer la production de lactate induite par la metformine comme moyen d’améliorer potentiellement l’efficacité thérapeutique. Dans cette étude, nous avons découvert que le DCA contribue à atténuer la production de lactate induite par la metformine et que le DCA y parvient par l’inhibition de PDK1.
Nos données démontrent que le DCA et la metformine induisent en synergie l’apoptose à un taux plus élevé que le traitement par la metformine seule. Nos données démontrent également que le DCA détourne le pyruvate vers une utilisation dans la phosphorylation oxydative, ce qui entraîne une accumulation de ROS dans le contexte de l’inhibition du complexe I par la metformine. Nous concluons que cette augmentation de la production de ROS conduit à des taux d’apoptose plus rapides.
Dans nos travaux précédents, nous avons démontré que la metformine induisait non seulement une mort cellulaire apoptotique caspase-dépendante, mais aussi une mort cellulaire PARP-dépendante survenant plus tard, qui a été révélée lorsqu’un inhibiteur pan-caspase a été utilisé [20]. Cette forme de mort cellulaire était associée à des changements morphologiques dans les mitochondries. Dans cette étude, le DCA a empêché les changements morphologiques mitochondriaux autrement induits par la metformine, tandis que le co-traitement a également conduit à un taux plus rapide de mort cellulaire. En outre, l’inhibition de la pan-caspase a presque complètement empêché la mort cellulaire et éliminé le clivage PARP associé au co-traitement par la metformine et le DCA. Tous ces éléments indiquent que le mécanisme de mort cellulaire induit par le traitement simultané est l’apoptose dépendante des caspases, et que l’apoptose est accélérée par rapport à la metformine seule. Il est possible que ce processus apoptotique soit trop catastrophique et ne nécessite pas ou ne laisse pas le temps de réaliser l’élargissement des mitochondries associé à la mort cellulaire PARP-dépendante de la metformine seule.
Au cours de la préparation de ce manuscrit, une étude de Choi et Lim [39] a été publiée, qui explorait le co-traitement du DCA et de la metformine dans les cellules cancéreuses. Nos résultats confirment et élargissent considérablement les conclusions de cette étude. Alors que l’étude de Choi et Lim se concentrait presque entièrement sur les cellules HeLa, notre étude démontre l’efficacité de l’association DCA et metformine sur plusieurs lignées cellulaires de cancer du sein. Il est important de noter que nous montrons également que cette combinaison de médicaments ne tue pas les cellules épithéliales mammaires non transformées, MCF10A, dans les mêmes conditions que celles dans lesquelles les médicaments tuent les cellules cancéreuses. Nous avons effectué des titrages minutieux qui démontrent une très forte synergie des médicaments dans l’induction de la mort cellulaire des cellules cancéreuses du sein. Ces résultats indiquent que le DCA sera efficace à des concentrations qui peuvent être atteintes de manière thérapeutique chez l’homme [27, 29], et à des niveaux bien inférieurs à ceux utilisés dans les expériences de l’étude de Choi et Lim (10-20 mM) [39]. Idéalement, la dose maximale de DCA utilisée pour traiter les cellules cancéreuses devrait être suffisamment faible pour éviter les effets indésirables tels que la neuropathie. Dans une étude de Michelakis et al. les taux sériques minimaux de DCA atteints étaient de 0,5 mM chez des patients atteints de glioblastome prenant 6,25 mg/kg par voie orale deux fois par jour [28]. Des doses plus élevées, comme 25 mg/kg, sont associées à une neuropathie liée à la dose. Par conséquent, les concentrations cliniquement pertinentes sont susceptibles de se situer dans la fourchette de 0,5 à 5 mM.
Les résultats présentés ici et ceux de Choi et Lim [39] ont montré que la mort cellulaire causée par la combinaison du DCA et de la metformine est associée au stress oxydatif. Nous montrons en outre que ce phénomène est associé à la production de superoxyde par les mitochondries induite par la metformine. Le dichloroacétate augmente encore la production de superoxyde mitochondrial en présence de la metformine, ce qui entraîne des dommages oxydatifs aux mitochondries, aux lipides cellulaires et à l’ADN. De nouvelles combinaisons telles que la metformine et le DCA sont prometteuses pour élargir les thérapies du cancer du sein.
Remerciements
Cet article a été soutenu par des subventions de Susan G. Komen for the Cure (KG100497) et du National Cancer Institute of the National Institutes of Health (1R01CA180033). Les noyaux de cytométrie en flux et d’imagerie ont été soutenus par les subventions COBRE P20GM103548 et P20GM103620 de l’Institut national des sciences médicales générales des Instituts nationaux de la santé.
Conflit d’intérêt
Les auteurs déclarent ne pas avoir de conflit d’intérêts.
Normes éthiques
Les auteurs déclarent que les expériences sont conformes aux lois en vigueur dans le pays où elles ont été réalisées.
RÉFÉRENCES
1 Warburg O (1956) On the origin of cancer cells. Science 123(80):309-314. doi:10.1016/S0306-9877(96)90136-X
2 DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB (2008) The biology of cancer : metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7:11-20. doi:10.1016/j.cmet.2007.10.002
3 Zamzami N, Kroemer G (2001) The mitochondrion in apoptosis : how Pandora’ s box opens. Nat Rev Mol Cell Biol 2:67-71. doi:10.1038/35048073
4 Gottfried E, Kunz-Schughart LA, Ebner S et al (2006) Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 107:2013-2021. doi:10.1182/blood-2005-05-1795
5 Fischer K, Hoffmann P, Voelkl S et al (2007) Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 109:3812-3819. doi:10.1182/blood-2006-07-035972
6 Jiralerspong S, Palla SL, Giordano SH et al (2009) Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J Clin Oncol 27:3297-3302. doi:10.1200/JCO.2009.19.6410
7 Evans JMM, Donnelly LA, Emslie-Smith AM et al (2005) Metformin and reduced risk of cancer in diabetic patients. BMJ 330:1303-1304. doi:10.1136/bmj.38393.572188.EB
8 Libby G, Donnelly LA, Donnan PT et al (2009) New users of Metformin are at low risk of incident cancer. Diabetes Care 32:1620-1625. doi:10.2337/dc08-2175
9 Zakikhani M, Dowling R, Fantus IG et al (2006) Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res 66:10269-10273. doi:10.1158/0008-5472.CAN-06-1500
10 Isakovic A, Harhaji L, Stevanovic D et al (2007) Dual antiglioma action of metformin : cell cycle arrest and mitochondria-dependent apoptosis. Cell Mol Life Sci 64:1290-1302. doi:10.1007/s00018-007-7080-4
11 Hadad SM, Appleyard V, Thompson AM (2008) L’activation thérapeutique de la metformine/AMPK favorise le phénotype angiogénique dans le modèle de cancer du sein ERα négatif MDA-MB-435. Breast Cancer Res Treat 114:391. doi:10.1007/s10549-008-0016-3
12 Buzzai M, Jones RG, Amaravadi RK et al (2007) Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res 67:6745-6752. doi:10.1158/0008-5472.CAN-06-4447
13 Sarbassov DD, Ali SM, Sabatini DM (2005) Growing roles for the mTOR pathway. Curr Opin Cell Biol 17:596-603. doi:10.1016/j.ceb.2005.09.009
14 Zhuang Y, Miskimins WK (2008) L’arrêt du cycle cellulaire dans les cellules cancéreuses du sein traitées à la Metformine implique l’activation de l’AMPK, la régulation négative de la cycline D1, et nécessite p27Kip1 ou p21Cip1. J Mol Signal 3:1-11. doi:10.1186/1750-2187-3-18
15 Hinke SA, Martens GA, Cai Y et al (2007) Methyl succinate antagonises biguanide-induced AMPK-activation and death of pancreatic beta-cells through restoration of mitochondrial electron transfer. Br J Pharmacol 150:1031-1043. doi:10.1038/sj.bjp.0707189
16 Zou M-H, Kirkpatrick SS, Davis BJ et al (2004) Activation of the AMP-activated protein kinase by the anti-diabetic drug metformin in vivo. Role of mitochondrial reactive nitrogen species. J Biol Chem 279:43940-43951. doi:10.1074/jbc.M404421200
17 Owen MR, Doran E, Halestrap AP (2000) Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 614:607-614. doi:10.1042/0264-6021:3480607
18 Carvalho C, Correia S, Santos MS et al (2008) Metformin promotes isolated rat liver mitochondria impairment. Mol Cell Biochem 308:75-83. doi:10.1007/s11010-007-9614-3
19d Kefas BA, Cai Y, Kerckhofs K et al (2004) Metformin-induced stimulation of AMP-activated protein kinase in beta-cells impairs their glucose responsiveness and can lead to apoptosis. Biochem Pharmacol 68:409-416. doi:10.1016/j.bcp.2004.04.003
20 Zhuang Y, Miskimins WK (2012) Metformin induce both caspase-dependent and poly(ADP-ribose) polymerase-dependent cell death in breast cancer cells. Mol Cancer Res 9:603-615. doi:10.1158/1541-7786.MCR-10-0343.Metformin
21 Bailey CJ, Wilcock C, Day C (1992) Effect of metformin on glucose metabolism in the splanchnic bed. Br J Pharmacol 105:1009-1013
22StacpoolePW, Lorenz AC, Thomas RG, Harman EM (1988) Dichloroacetate in the treatment of lactic acidosis. Ann Intern Med 108:58-63. doi:10.1056/NEJM198308183090702
23 Whitehouse BSUE, Cooper RH, Randle PJ (1974) Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem J 141:761-774. doi:10.1056/NEJM198308183090702
24 Hussien R, Brooks GA (2011) Expression des isoformes du transporteur de lactate et de la lactate déshydrogénase de la membrane mitochondriale et plasmique dans les lignées cellulaires du cancer du sein. Physiol Genomics 43:255-264. doi:10.1152/physiolgenomics.00177.2010
25 Kim J, Tchernyshyov I, Semenza GL, Dang CV (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase : a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3:177-185. doi:10.1016/j.cmet.2006.02.002
26 Wigfield SM, Winter SC, Giatromanolaki A et al (2008) PDK-1 régule la production de lactate dans l’hypoxie et est associé à un mauvais pronostic dans le cancer squameux de la tête et du cou. Br J Cancer 98:1975-1984. doi:10.1038/sj.bjc.6604356
27 Stacpoole PW, Kurtz TL, Han Z, Langaee T (2008) Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv Drug Deliv Rev 60:1478-1487. doi:10.1016/j.addr.2008.02.014
28 Michelakis ED, Sutendra G, Dromparis P et al (2010) Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2:31ra34. doi:10.1126/scitranslmed.3000677
29 Mori M, Yamagata T, Goto T et al (2004) Dichloroacetate treatment for mitochondrial cytopathy : long-term effects in MELAS. Brain Develop 26:453-458. doi:10.1016/j.braindev.2003.12.009
30 Chou T (2010) drug combination studies and their synergy quantification using the Chou-Talalay Method drug combination studies and their synergy quantification using the Chou-Talalay Method. Cancer Res 70:440-446. doi:10.1158/0008-5472.CAN-09-1947
31 Mah L, Karagiannis TC (2010) γH2AX : a sensitive molecular marker of DNA damage and repair. Leukemia 24:679-686. doi:10.1038/leu.2010.6
32 Alimova IN, Liu B, Fan Z et al (2009) La metformine inhibe la croissance des cellules du cancer du sein, la formation de colonies et induit un arrêt du cycle cellulaire in vitro. Cell Cycle 8:909-915. doi:7933 [pii]
33 Soini Y, Vakkala M, Kahlos K et al (2001) L’expression de la MnSOD est moins fréquente dans les cellules tumorales des carcinomes mammaires invasifs que dans les carcinomes in situ ou les cellules épithéliales mammaires non néoplasiques. J Pathol 195:156-162. doi:10.1002/path.946
34 Walenta S, Wetterling M, Lehrke M et al (2000) High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers high lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical. Cancer Res 60:916-921
35 Leek R, Harris AL (2009) Lactate dehydrogenase 5 expression in squamous cell head and neck cancer relates to prognosis following radical or postoperative radiotherapy. Oncology 77:285-292. doi:10.1159/000259260
36 Isidoro A, Casado E, Redondo A et al (2005) Breast carcinomas fulfill the Warburg hypothesis and provide metabolic markers of cancer prognosis. Carcinogenèse 26:2095-2104. doi:10.1093/carcin/bgi188
37 Isidoro A, Martínez M, Fernández PL et al (2004) Alteration of the bioenergetic phenotype of mitochondria is a hallmark of breast, gastric, lung and oesophageal cancer. Biochem J 378:17-20. doi:10.1042/BJ20031541
38 Walenta S, Schroeder T (2004) Lactate in solid malignant tumors : potential basis of a metabolic classification in clinical oncology. Curr Med Chem 11:2195-2204
39 Choi YW, Lim IK (2014) Sensitization of metformin-cytotoxicity by dichloroacetate via reprogramming glucose metabolism in cancer cells. Cancer Lett 346:300-308. doi:10.1016/j.canlet.2014.01.015
Contenu connexe :