Cecilie Abildgaard1, Christina Dahl1, Ahmad Abdul-Al1, Annette Christensen1 y Per Guldberg1
1 Centro de Investigación de la Sociedad Danesa del Cáncer, Copenhague, Dinamarca
Correspondencia: Per Guldberg, correo electrónico: [email protected]
Recibido: 19 de abril de 2017
Aceptado: 19 de julio de 2017
Publicado: 24 de agosto de 2017
Resumen
La desregulación del metabolismo durante la progresión del melanoma está estrechamente asociada con la adquisición de alteraciones genéticas y epigenéticas en los reguladores de las vías metabólicas. El receptor beta del ácido retinoico (RARβ) está silenciado epigenéticamente en una gran proporción de melanomas, pero no se ha establecido un vínculo entre RARβ y el recableado metabólico del melanoma. Aquí demostramos que, en melanocitos humanos primarios, el ácido retinoico todo trans (un agonista de RARβ) induce una inhibición del crecimiento acompañada de una disminución del metabolismo glucolítico y oxidativo, mientras que la inhibición selectiva de RARβ conduce a un aumento de la tasa glucolítica basal y a una mayor sensibilidad a la inhibición de la glucólisis. En las células de melanoma, la inhibición de RARβ promovió una menor respiración mitocondrial y una mayor actividad glucolítica, lo que provocó estrés energético y la activación del sensor energético proteína quinasa activada por AMP. Este cambio metabólico aumentó la sensibilidad tanto a la inhibición glucolítica como a la estimulación del metabolismo mitocondrial con dicloroacetato, un inhibidor de la piruvato deshidrogenasa cinasa. En células de melanoma portadoras de la mutación BRAFV600E, la activación de RARβ antagonizó el efecto del inhibidor de BRAF PLX4032 (vemurafenib). En conjunto, estos datos sugieren que la señalización RARβ interviene en la regulación del metabolismo celular en el melanoma y puede constituir una posible diana en las estrategias de tratamiento combinado.
Palabrasclave: melanoma, metabolismo del cáncer, receptor β del ácido retinoico, respiración mitocondrial, dicloroacetato
Abreviaturas: ATRA: ácido retinoico todo trans; DCA: dicloroacetato; ECAR: tasa de acidificación extracelular; OCR: tasa de consumo de oxígeno; ROS: especies reactivas del oxígeno
© Abildgaard et al. Este es un artículo de acceso abierto distribuido bajo los términos de la Creative Commons Attribution License 3.0 (CC BY 3.0), que permite el uso, distribución y reproducción sin restricciones en cualquier medio, siempre que se cite al autor original y la fuente.
INTRODUCCIÓN
El melanoma, la forma más letal de cáncer de piel, causa 50.000 muertes al año y su incidencia sigue aumentando en todo el mundo. Aunque el melanoma cutáneo primario es curable mediante cirugía, la forma más avanzada de la enfermedad (estadio IV) se asocia a una supervivencia a 10 años del 10-15% [1], lo que refleja su notoria resistencia a la terapia anticancerosa convencional. Los avances terapéuticos recientes incluyen inhibidores de puntos de control inmunitarios y terapias dirigidas a oncogenes o efectores descendentes de la vía MAPK (por ejemplo, inhibidores de BRAF y MEK). Sin embargo, el desarrollo de resistencia farmacológica adquirida acaba provocando una recaída en la mayoría de los casos [2, 3].
El melanoma se desarrolla a partir de células productoras de melanina, denominadas melanocitos, mediante la adquisición de múltiples alteraciones genómicas. Los impulsores más comunes del melanoma incluyen mutaciones activadoras en BRAF y NRAS y mutaciones inactivadoras o deleciones en CDKN2A (que codifica p16INK4A y p14ARF), PTEN y TP53 [4]. Pruebas recientes sugieren que una función común que comparten algunos de estos genes es el control del metabolismo celular [5, 6]. Durante la progresión del melanoma, el metabolismo celular se reprograma, lo que implica un cambio de la respiración mitocondrial hacia la glucólisis aeróbica, que conduce a un aumento del consumo de glucosa y de la producción de ácido láctico (efecto Warburg) [7]. Varios informes basados en modelos in vitro e in vivo de melanoma y estudios clínicos de pacientes con melanoma han demostrado una relación entre las mutaciones activadoras en el codón V600 de BRAF (más comúnmente BRAFV600E) y la glucólisis aeróbica [8-10]. A nivel molecular, BRAFV600E regula la fosforilación oxidativa mediante la supresión del regulador maestro de la biogénesis mitocondrial, PGC1α, a través de la inhibición del factor de transcripción asociado a la microftalmia (MITF). Por el contrario, la inhibición de BRAFV600E conduce a la adicción oxidativa a través de la inducción de PGC1α y el aumento de la respiración mitocondrial [11]. La correspondiente disminución de la actividad glucolítica puede visualizarse mediante PET-TAC en pacientes con melanoma tratados con inhibidores de BRAF, mostrando una menor captación de glucosa en el tejido tumoral [10]. Los ensayos clínicos de fase III del inhibidor de BRAFV600E vemurafenib (PLX4032) demostraron una mejora de la supervivencia global y libre de progresión en pacientes con melanoma metastásico [12]. Se ha sugerido que los inhibidores mitocondriales son útiles como adyuvantes de los inhibidores de la vía BRAF para mejorar el efecto o prevenir el desarrollo de farmacorresistencia [13-15].
Además de los factores genéticos bien caracterizados, el genoma del melanoma contiene numerosas alteraciones epigenéticas. Una de las dianas epigenéticas recurrentes en el melanoma es RARB, que codifica el receptor beta del ácido retinoico (RARβ), silenciado por hipermetilación del promotor en el 45-70% de los melanomas cutáneos [16, 17]. En las células del linaje melanocítico, RARβ media en la inhibición del crecimiento y la melanogénesis inducida por el ácido retinoico (vitamina A), un marcador de la diferenciación melanocítica [18]. Anteriormente hemos demostrado que la activación de RARβ en melanocitos induce la regulación al alza de p14ARF [17], que protege contra la disfunción mitocondrial y el estrés oxidativo [19]. Aquí mostramos que los melanocitos humanos responden a la activación de RARβ reduciendo el metabolismo oxidativo, potencialmente como parte de una respuesta de diferenciación. En células de melanoma, la activación de RARβ antagoniza el efecto de PLX4032, mientras que la inhibición de RARβ induce dependencia glucolítica y estrés energético, haciendo que las células sean vulnerables al tratamiento con el inhibidor de la piruvato deshidrogenasa cinasa dicloroacetato (DCA).
RESULTADOS
La activación de RARβ reduce el crecimiento y la tasa metabólica de los melanocitos
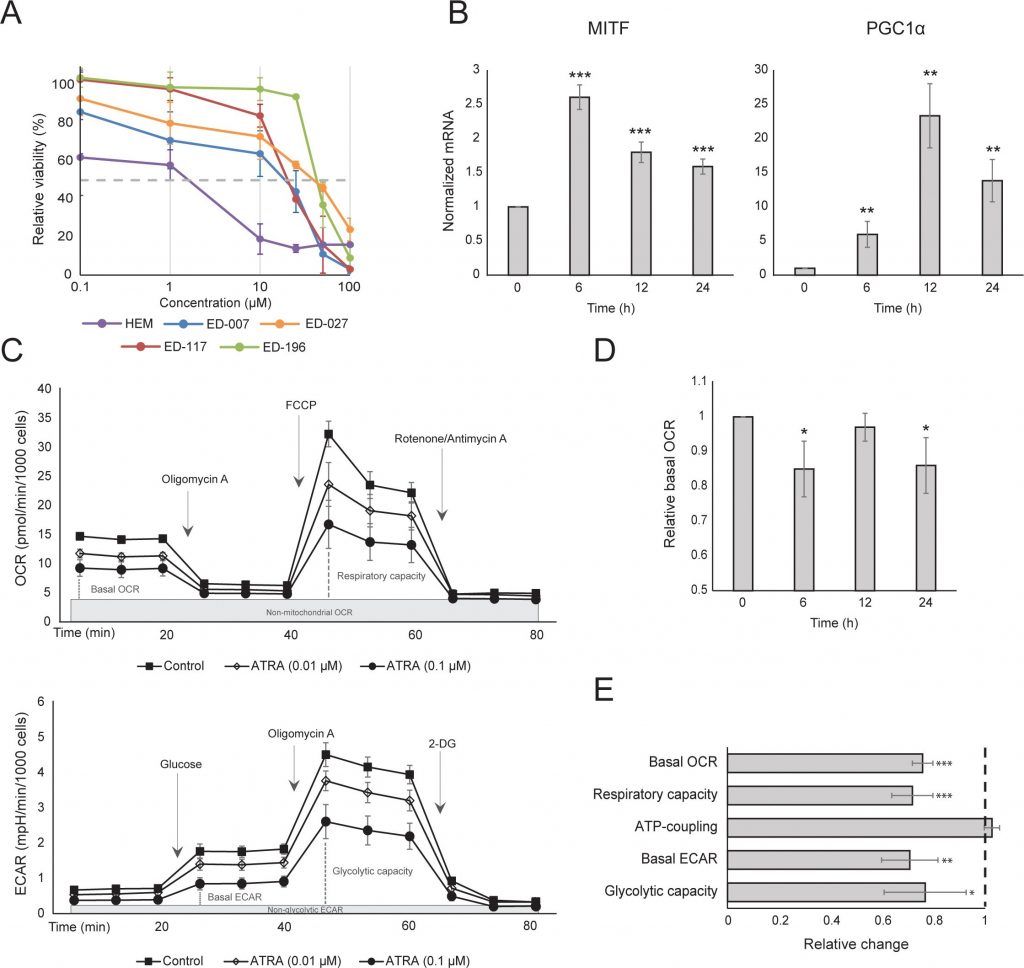
En primer lugar, determinamos el efecto de la activación de RARβ sobre el crecimiento de melanocitos epidérmicos humanos primarios. Las células se trataron con el agonista RARβ ácido transretinoico total (ATRA) durante 6 días, y la respuesta de crecimiento se determinó con un ensayo de viabilidad basado en violeta de cristal. En consonancia con informes anteriores [17, 20, 21], el ATRA redujo el crecimiento de melanocitos de forma dosis-dependiente (Figura 1A), con un IC50 de 2,4 μM (Tabla 1). Se ha demostrado previamente que el tratamiento a corto plazo (<24 h) con atra induce la diferenciación y la melanogénesis en los melanocitos, mientras que la exposición a largo plazo (>24 h) reduce la proliferación e induce la apoptosis [20,
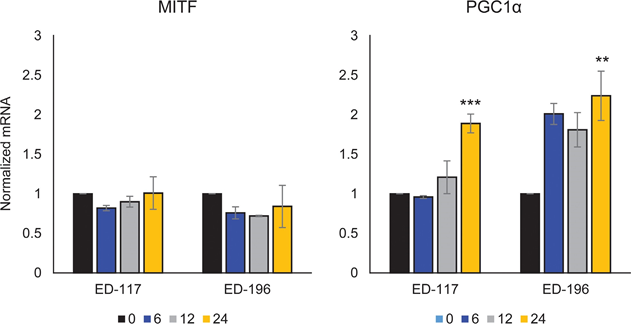
]. Descubrimos que el ATRA (0,1 μM) inducía un aumento transitorio del factor de transcripción específico del linaje melanocítico MITF (factor de transcripción asociado a la microftalmia), con un pico de expresión a las 6 h y un posterior descenso hacia los niveles basales (Figura 1B). En las células de melanoma, MITF regula la expresión de PGC1α, un marcador de fenotipo oxidativo [22]. Por lo tanto, investigamos la expresión de PGC1α en melanocitos en diferentes puntos temporales tras la exposición a ATRA (0,1 μM). Como se muestra en la Figura 1B, PGC1α también se incrementó transitoriamente, con un retraso de ~6 h en relación con MITF.
| Células/líneas celulares | Células/líneas celulares | Características | Características | Características | Valores IC50 | Valores IC50 | Valores IC50 | Valores IC50 |
|---|---|---|---|---|---|---|---|---|
| Número ED | Nombre | Estado de BRAF* | Expresión de RARβ ** | expresión de p14ARF | ATRA (μM) | LE135 (μM) | DCA (mM)*** | PLX4032 (μM) |
| HEM | WT | + | + | 2.4±1.6 | 2.8±0.8 | 69.1±6.4 | NA | |
| ED-007 | FM-3 | WT | + | – | 18.6±8.7 | 8.6±1.0 | 12.2±2.2 | NA |
| ED-027 | FM-82 | BRAFV600E | + | + | 39.8±5.3 | 10.7±1.3 | 17.7±2.1 | 0.52±0.04 |
| ED-117 | Mel-NT3-00 | BRAFV600E | + | + | 25.5±5.0 | NA | 37.6 ±2.2 | 0.51±0.09 |
| ED-196 | Ma-Mel-51 | BRAFV600E | + | + | 46.2±9.1 | 8.4±0.4 | 35.8±3.2 | 0.26±0.06 |
Los valores IC50 representan la media ± desviación estándar de ≥3 experimentos independientes.
*Confirmadoporpirosecuenciación
**ConfirmadoporqPCR
***Valores IC50publicadospor Abildgaard et al. [29]
#Melanocitos epidérmicos humanos
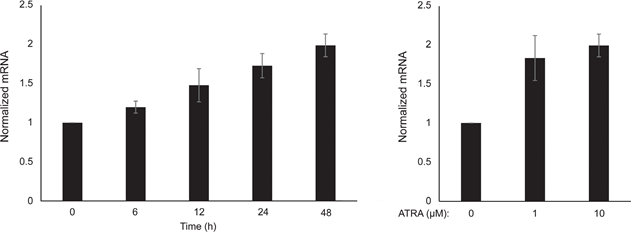
Debido al papel de PGC1α en la biogénesis mitocondrial, investigamos a continuación si la expresión de PGC1α se correlacionaba con el nivel de respiración mitocondrial. Utilizando el instrumento Seahorse XFe96, medimos la tasa de consumo de oxígeno (OCR) y la tasa de acidificación extracelular (ECAR), que son indicadores de la tasa respiratoria mitocondrial y de la actividad glucolítica, respectivamente. La OCR y la ECAR se midieron durante la adición secuencial de moduladores metabólicos, lo que permitió determinar las tasas y capacidades basales de los dos sistemas energéticos (Figura 1C). Para comprender mejor la dependencia temporal de las respuestas al ATRA, se midieron los parámetros metabólicos tras exposiciones a corto plazo (6-24 h) y a largo plazo (7 días). Tras el tratamiento de los melanocitos con ATRA (0,1 μM) durante 6 o 24 h, se redujo el OCR basal. Sin embargo, tras 12 h de tratamiento, el OCR fue similar a los niveles basales (Figura 1D). Estas fluctuaciones en el estado metabólico coincidieron con cambios en la expresión de MITF y PGC1α. La exposición prolongada (7 días) a una dosis baja de ATRA (0,01 μM) produjo una disminución adicional tanto de la OCR basal como de la capacidad respiratoria (Figura 1E).
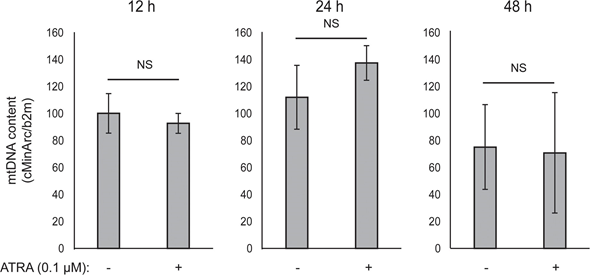
El acoplamiento mitocondrial de ATP no se vio afectado por el ATRA (Figura 1E), resultando en una disminución neta de la producción de ATP de las mitocondrias. Se ha demostrado que PGC1α regula al alza la proteína 2 de desacoplamiento (UCP2), lo que conduce a un ligero desacoplamiento mitocondrial [23, 24]. La expresión de UCP2 en melanocitos no se vio afectada por el tratamiento con ATRA (Figura Suplementaria 1), lo que apoya aún más que el acoplamiento ATP se mantuvo inalterado durante la activación RARβ. La expresión de marcadores de actividad mitocondrial (COX5A, ATP5g1 y NDUFS3) y el contenido de ADN mitocondrial tampoco se vieron afectados por el tratamiento con ATRA (0,1 μM) hasta 24 y 48 h, respectivamente (Figuras suplementarias 1 y 2).
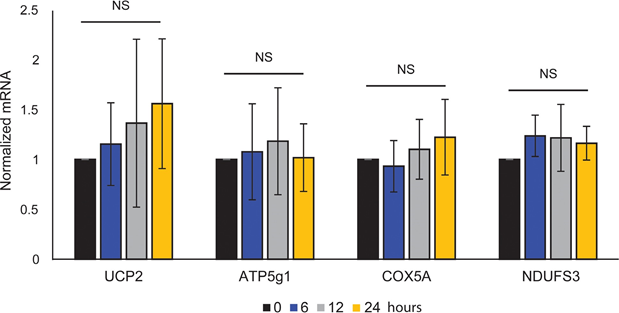
no mostró significación estadística (NS).
No hubo cambios en la tasa glucolítica después de 24 h de tratamiento con ATRA (datos no mostrados); sin embargo, después de 7 días, la actividad glucolítica basal y la capacidad glucolítica se redujeron significativamente (Figura 1E). La supresión de los dos principales sistemas energéticos celulares indica que los melanocitos presentan una menor demanda energética en presencia de ATRA, lo que podría ser consecuencia de un menor crecimiento celular.
La inhibición de RARβ aumenta la tasa glucolítica basal y promueve la dependencia glucolítica en melanocitos
Unretoa la hora de estudiar los efectos celulares del ATRA es la presencia de concentraciones desconocidas de vitamina A en el suero bovino fetal, una fuente esencial de micronutrientes en la mayoría de los medios de cultivocelular
[25]. Para estudiar con más detalle el papel de la señalización RARβ en el metabolismo de los melanocitos, utilizamos el antagonista de RARβ LE135, que se dirige a RARβ con una selectividad moderada sobre RARα y alta sobre RARγ y RXRα [26].
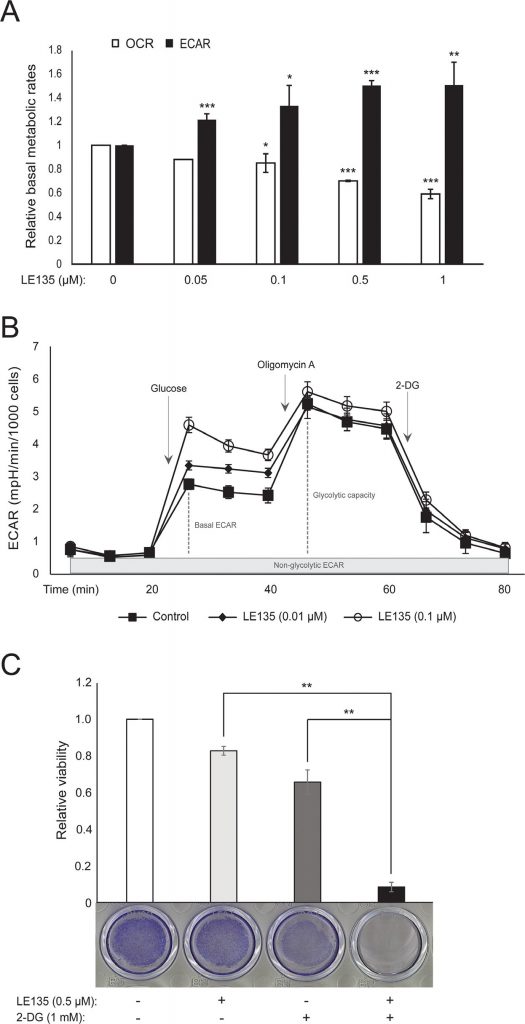
Repetimos los protocolos Seahorse mostrados en la Figura 1C en melanocitos tratados con diferentes concentraciones de LE135 durante 24 h y 7 días. Tras 24 h, observamos un aumento dependiente de la dosis de la actividad glucolítica, con un aumento del ECAR basal de hasta el 50%, y una reducción correspondiente del OCR (Figura 2A). El ECAR basal seguía aumentado tras 7 días de tratamiento (Figura 2B). No se produjo un aumento significativo de la capacidad glucolítica, lo que sugiere que las células se vieron obligadas a depender de la glucólisis para la producción de energía. Esto se vio corroborado por una mayor sensibilidad de estas células al inhibidor de la glucólisis 2-deoxi-D-glucosa (2-DG), en presencia de LE135 (Figura 2C).
ATRA antagoniza el efecto de la inhibición de BRAF en células de melanoma
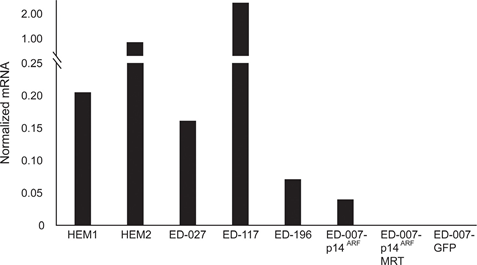
RARB está silenciado por hipermetilación del promotor en muchos melanomas, lo que sugiere que posee propiedades supresoras de tumores [16]. En un estudio previo de eventos genéticos y epigenéticos en 110 líneas celulares de melanoma, encontramos una prevalencia del 66% para las mutaciones de BRAF y del 45% para la hipermetilación del promotor de RARB, sin correlación entre estos dos eventos [17]. Para ampliar estos datos, examinamos la expresión de RARβ en 84 de estas líneas celulares de melanoma, así como en melanocitos epidérmicos humanos. Los niveles de expresión variaron enormemente entre las líneas celulares de melanoma, desde la ausencia total de expresión hasta niveles hasta 27 veces superiores a los melanocitos (Figura 3A). Como era de esperar, la hipermetilación del promotor de RARB se asoció con niveles de expresión de RARβ de bajos a indetectables, con pocas excepciones. No hubo asociación entre las mutaciones BRAFV600E y los niveles de expresión de RARβ (Figura 3A), lo que sugiere que la sensibilidad de las células de melanoma al ATRA puede ser independiente del estado de BRAF.
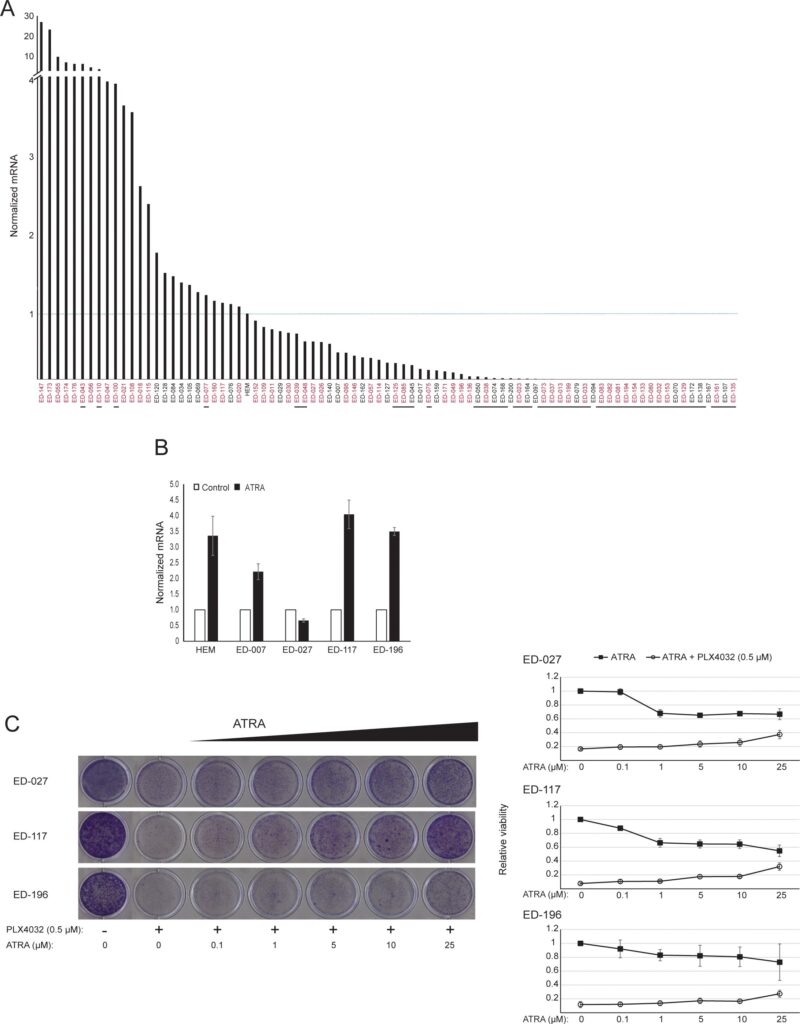
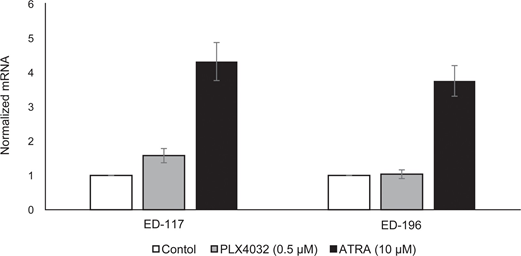
Para seguir estudiando el papel de la señalización RARβ en el melanoma, seleccionamos cuatro líneas celulares de melanoma RARβ-positivas (ED-007, ED-027, ED-117 y ED-196) para el análisis funcional. Tres de estas líneas celulares presentaban mutación BRAFV600E (ED-027, ED-117 y ED-196) y una era de tipo salvaje BRAF (ED-007). El tratamiento con ATRA provocó una disminución del crecimiento de las cuatro líneas celulares (Figura 1A), aunque su sensibilidad fue menor que la de los melanocitos, indicada por los valores de IC50 (Tabla 1). Se sabe que la expresión de RARβ se induce en respuesta al ATRA [27]. Como se muestra en la Figura 3B, RARβ se indujo en 3 de las cuatro líneas celulares de melanoma. A pesar de una inducción de RARβ más pronunciada en ED-117 y ED-196, el efecto del ATRA sobre la expresión de MITF y PGC1α se atenuó en comparación con los melanocitos (Figura suplementaria 3). Anteriormente se demostró que la supresión del eje MITF/PGC1α era consecuencia de la actividad oncogénica de BRAF [11], lo que podría contribuir a una menor respuesta al ATRA. En consonancia con esta noción, las células de tipo salvaje BRAF mostraron la mayor sensibilidad al ATRA, aunque todavía considerablemente inferior a la de los melanocitos (Tabla 1).
Para investigar si el tratamiento de BRAF con PLX4032 restablecería la sensibilidad al ATRA, tratamos las líneas celulares de melanoma mutantes para BRAFV600E con PLX4032 a una concentración cercana a los valores IC50 (véase la Tabla 1) en combinación con concentraciones crecientes de ATRA (0,1-25 μM). Curiosamente, el ATRA rescató el efecto citotóxico de PLX4032 en todas las líneas celulares. El efecto dosis-dependiente del ATRA sobre el crecimiento de las células de melanoma tratadas con PLX4032 (Figura 3C) indica antagonismo entre los dos compuestos. El tratamiento con PLX4032 (0,5 μM) no redujo la expresión de RARβ (Figura suplementaria 4), lo que sugiere un mecanismo diferente para este antagonismo.
Elefecto del ATRA sobre el metabolismo celular se ve afectado por el estado de p14ARF
La observación de que tanto el ATRA como PLX4032 afectan a la biogénesis mitocondrial podría apuntar a una explicación metabólica del efecto antagónico de estos compuestos. Anteriormente se demostró que p14ARF se expresa como proteína citoplasmática en melanocitos normales y que protege a estas células frente a mitocondrias disfuncionales [19]. De acuerdo con hallazgos previos [17], el ATRA aumentó la expresión de p14ARF en células de melanoma RARβ-positivas (Figura Suplementaria 5). Paradójicamente, aunque p14ARF se pierde con frecuencia en el melanoma a través de la deleción del locus CDKN2A, no es posible establemente knock down ARF en células de melanoma que expresan este gen [17]; y datos no mostrados). En su lugar, para estudiar el papel potencial de p14ARF en la mediación de una respuesta celular al ATRA, transfectamos de forma estable la línea celular de melanoma ED-007 deficiente en p14ARF con un constructo EGFP-p14ARF. La expresión de p14ARF en las células transfectadas se verificó mediante qPCR (Figura suplementaria 6). El análisis Seahorse mostró diferentes perfiles metabólicos (Figura 4A) con un OCR basal y una capacidad respiratoria significativamente menores en las células con expresión de p14ARF restaurada en comparación con las células transfectadas como control (Figura 4B). Curiosamente, las células con expresión de p14ARF también mostraron una mayor sensibilidad al ATRA (Figura 4C).
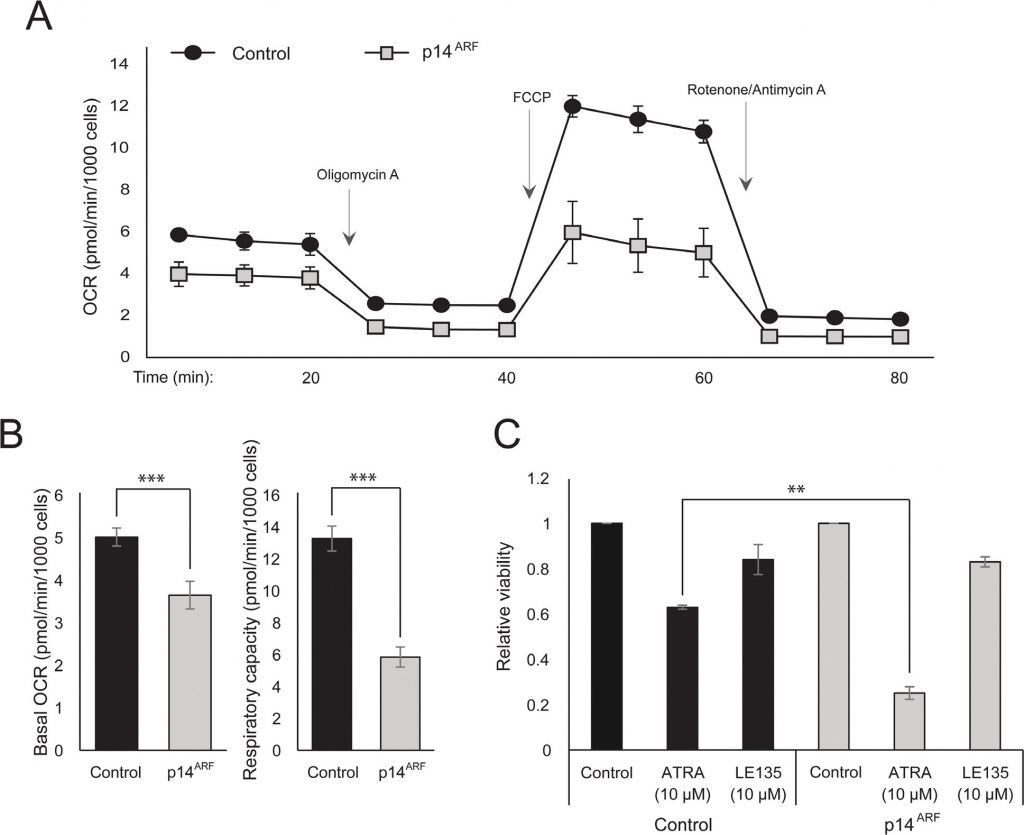
El bloqueo de RARβ induce dependencia glucolítica y estrés energético en células de melanoma y las sensibiliza al dicloroacetato
Basándonos en el hallazgo de un efecto antagonista entre PLX4032 y ATRA, probamos el efecto combinado de PLX4032 y LE135 en busca de un sinergismo potencial en células de melanoma ED-117 y ED-196. No se demostró ninguna inhibición cooperativa del crecimiento del melanoma en el montaje experimental utilizado aquí (6 días de tratamiento con PLX4032 [0,1 μM] y LE135 [1 μM]; Figura suplementaria 7).
Para estudiar más a fondo el efecto de la inhibición de RARβ en el metabolismo del melanoma, medimos OCR y ECAR en líneas celulares de melanoma RARβ-positivas tratadas con LE135. De forma similar a lo observado en melanocitos (Figura 2A-2B), LE135 aumentó la tasa glucolítica basal y redujo la respiración mitocondrial basal en células de melanoma (Figura 5A-5B). Este cambio metabólico, que es coherente con el efecto Warburg, fue más marcado en las células de melanoma en comparación con los melanocitos, lo que condujo a un aumento de la tasa glucolítica basal para alcanzar la capacidad máxima. Esto quedó ilustrado por la incapacidad de seguir aumentando la ECAR tras la adición de oligomicina A, lo que indica una falta de flexibilidad metabólica (Figura 5B). Para investigar el efecto de LE135 en la bioenergética celular, investigamos el estado de fosforilación de la proteína quinasa activada por AMP (AMPK). La AMPK detecta el estado energético celular reaccionando a niveles elevados de AMP, que se acumula a medida que disminuye la relación ATP/ADP. Así, la reducción de la producción de energía o el aumento del consumo energético pueden promover un aumento del AMP, que se une a la AMPK y conduce a su fosforilación y activación [28]. El tratamiento de líneas celulares de melanoma con LE135 provocó la fosforilación de AMPK, lo que indica que las células están sometidas a estrés energético. El aumento de los niveles de p-AMPK fue evidente ya después de 2 h y siguió aumentando hasta al menos 48 h (Figura 5C). Estos resultados fueron diferentes de los de los melanocitos, en los que el tratamiento con LE135 no provocó la activación de AMPK (Figura suplementaria 8). Tanto las células de melanoma como los melanocitos mostraron una reducción del crecimiento celular durante 6 días de tratamiento con LE135 (los valores IC50 se muestran en la Tabla 1). Además, de forma similar a los melanocitos, LE135 sensibilizó las células de melanoma a la inhibición glucolítica con 2-DG (Figura 5D).
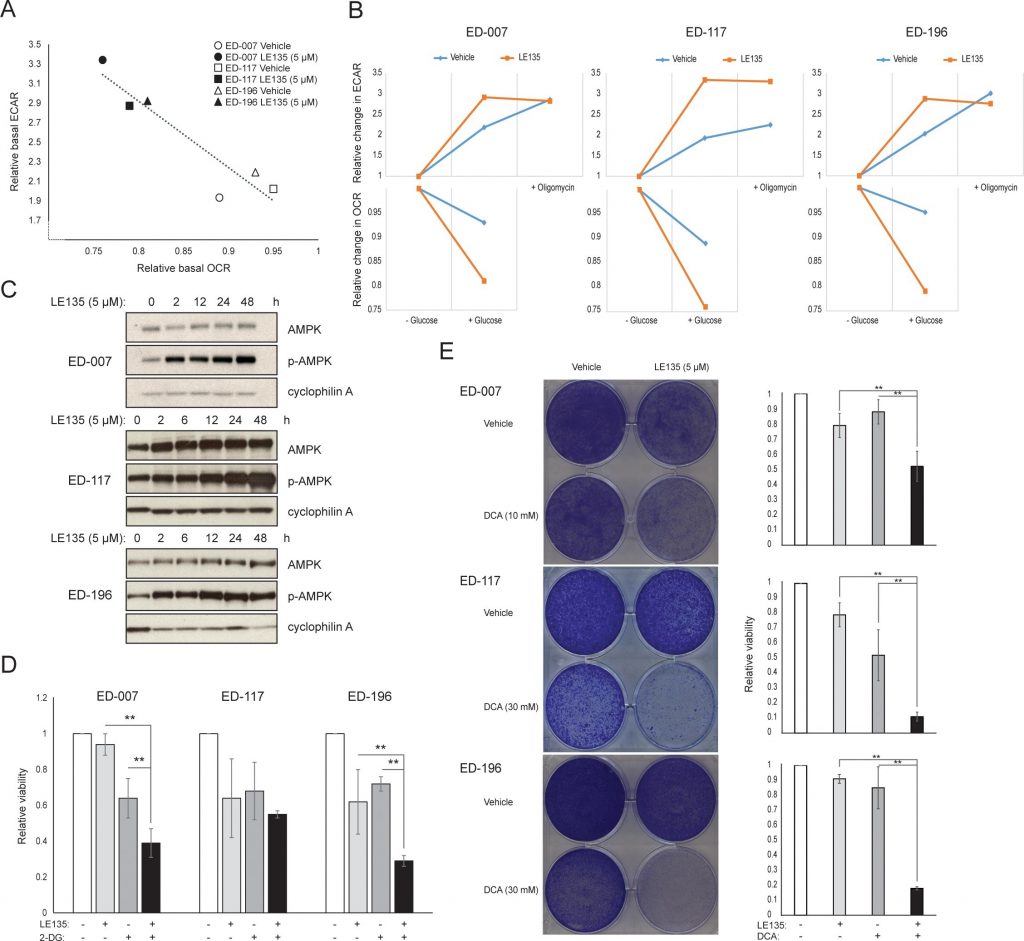
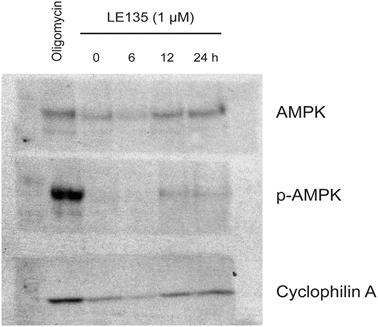
La inducción de estrés energético con LE135 en células de melanoma pero no en melanocitos alude a una potencial relevancia terapéutica de este compuesto en estrategias de tratamiento combinado. Se ha demostrado previamente que el inhibidor de la piruvato deshidrogenasa cinasa DCA inhibe el crecimiento de células de melanoma al inducir un cambio en el metabolismo alejándolo de la glucólisis, haciendo que las células dependan de la respiración mitocondrial [9,29-32]. Además, se ha demostrado que el DCA inhibe el crecimiento de diversas líneas celulares de melanoma, independientemente del estado de BRAF y de la sensibilidad a PLX4032 [29]. Para estudiar el efecto combinado de LE135 y DCA, aplicamos concentraciones inferiores a los respectivos valores IC50 para cada una de las tres líneas celulares de melanoma (cf. Tabla 1). A pesar del escaso efecto sobre la reducción del crecimiento de cada compuesto por separado (9-21% para LE135 y 12-48% para DCA), la combinación indujo una reducción de hasta el 89% (Figura 5E). Estos resultados sugieren que los efectos opuestos de LE135 (que promueve la dependencia glucolítica) y DCA (que aleja a las células de la glucólisis) pueden actuar sinérgicamente para inhibir el crecimiento del melanoma.
DISCUSIÓN
El ATRA y otros derivados de la vitamina A reducen el crecimiento celular e inducen la expresión de marcadores de diferenciación en diversos tejidos [33, 34]. Hemos observado que, en melanocitos humanos primarios, el ATRA induce un aumento transitorio del eje MITF/PGC1α, en consonancia con el aumento de la función mitocondrial inducido por el ATRA observado en otros tipos celulares como los adipocitos y los hepatocitos [35-37]. Sin embargo, el tratamiento a largo plazo con bajas concentraciones de ATRA condujo a reducciones en el crecimiento celular y en la tasa metabólica, determinadas por una menor glucólisis basal, así como por una menor respiración mitocondrial. Estos cambios metabólicos en respuesta al ATRA probablemente reflejan una respuesta de diferenciación hacia el estado no proliferativo que caracteriza a los melanocitos residentes en la piel. El bloqueo de la señalización RARβ en estas células provocó un aumento de la tasa glucolítica basal y la correspondiente disminución del metabolismo oxidativo. La ventaja selectiva de perder la función de RARβ en el melanoma, por ejemplo mediante la hipermetilación de RARB, podría estar relacionada con la transición a un fenotipo más dependiente de la glucólisis que apoya el efecto Warburg.
Contrariamente a la situación en melanocitos primarios, el bloqueo de la señalización RARβ en células de melanoma provocó estrés energético, como indica la activación de AMPK. Esta respuesta podría ser el resultado de una menor capacidad de las células de melanoma para aumentar la glucólisis en comparación con los melanocitos. Mientras que los melanocitos tienen un nivel glucolítico basal relativamente bajo y pueden pasar a una actividad más elevada cuando lo necesitan, las células de melanoma se caracterizan por una elevada tasa glucolítica cercana a su capacidad máxima. Así, los melanocitos son más flexibles a la hora de adaptarse a la inhibición de la señalización RARβ para mantener el nivel energético. Esto indica que existe una ventana terapéutica relevante para LE135 y otros inhibidores de RARβ en terapias combinadas, por ejemplo con DCA. De forma similar a los efectos metabólicos de la inhibición de BRAF, el DCA desvía a las células cancerosas glucolíticas de la glucólisis hacia la respiración mitocondrial [9, 38, 39], pero a diferencia del PLX4032, el efecto del DCA no se limita a los melanomas con mutación de BRAF [29]. En un estudio anterior, demostramos que el cambio metabólico inducido por el DCA se correlacionaba con una reducción de los niveles de ATP, lo que sugería que el DCA podría estar dirigido a la homeostasis bioenergética de las células de melanoma [29]. En este estudio se observó que la combinación de LE135 y DCA atenuaba de forma cooperativa el crecimiento de células de melanoma que expresaban el receptor RARβ. El tratamiento de las células de melanoma con DCA o LE135 podría darles una ventana para adaptarse a las nuevas demandas metabólicas, mientras que la combinación de los tratamientos limitaría la flexibilidad metabólica y las haría incapaces de sostener la producción de energía necesaria para un crecimiento continuado.
En muchos melanomas, la expresión de PGC1α es baja debido a la supresión de MITF por mutaciones oncogénicas de BRAF [11]. Este fenotipo favorece el efecto Warburg al obligar a las células a cambiar a la glucólisis para la producción de energía. El tratamiento con inhibidores de BRAF restaura la expresión de PGC1α y cambia el patrón metabólico de nuevo hacia la respiración mitocondrial [
, 40]. El efecto citotóxico de la inhibición de BRAF puede verse potenciado por la presencia de mitocondrias disfuncionales en las células de melanoma, lo que resulta en un aumento de la producción de ROS [13, 41]. Descubrimos que el ATRA potencia el crecimiento de las células de melanoma en presencia del inhibidor de BRAF PLX4032. Basándonos en los conocimientos de estudios anteriores [
, 17, 19, 41] y en los hallazgos aquí presentados, proponemos un modelo que integra los efectos metabólicos de PLX4032 y ATRA y explica su antagonismo (Figura 6). El modelo sugiere un papel dual del aumento de la señalización RARβ, que conduce a la activación de PGC1α y la biogénesis mitocondrial, mientras que al mismo tiempo suprime la OCR y la producción de ROS a través de la inducción de p14ARF. El modelo se apoya en los resultados del presente estudio y de un estudio anterior [17] que muestran que el tratamiento con ATRA induce la expresión de p14ARF, y que la reconstitución de células de melanoma deficientes en p14ARF con p14ARF de tipo salvaje reduce la OCR y aumenta la sensibilidad al ATRA. Así pues, el ATRA podría reducir la sensibilidad de los melanomas RARβ-positivos al PLX4032 limitando la citotoxicidad de la producción de ROS. La vitamina A se ha propuesto con fines profilácticos y terapéuticos en muchos tipos de cáncer, incluido el melanoma [42]. Aunque falta la validación clínica, nuestros resultados desaconsejan el uso de suplementos de vitamina A en pacientes con melanoma sometidos a tratamiento con inhibidores de BRAF, debido al potencial efecto antagonista.
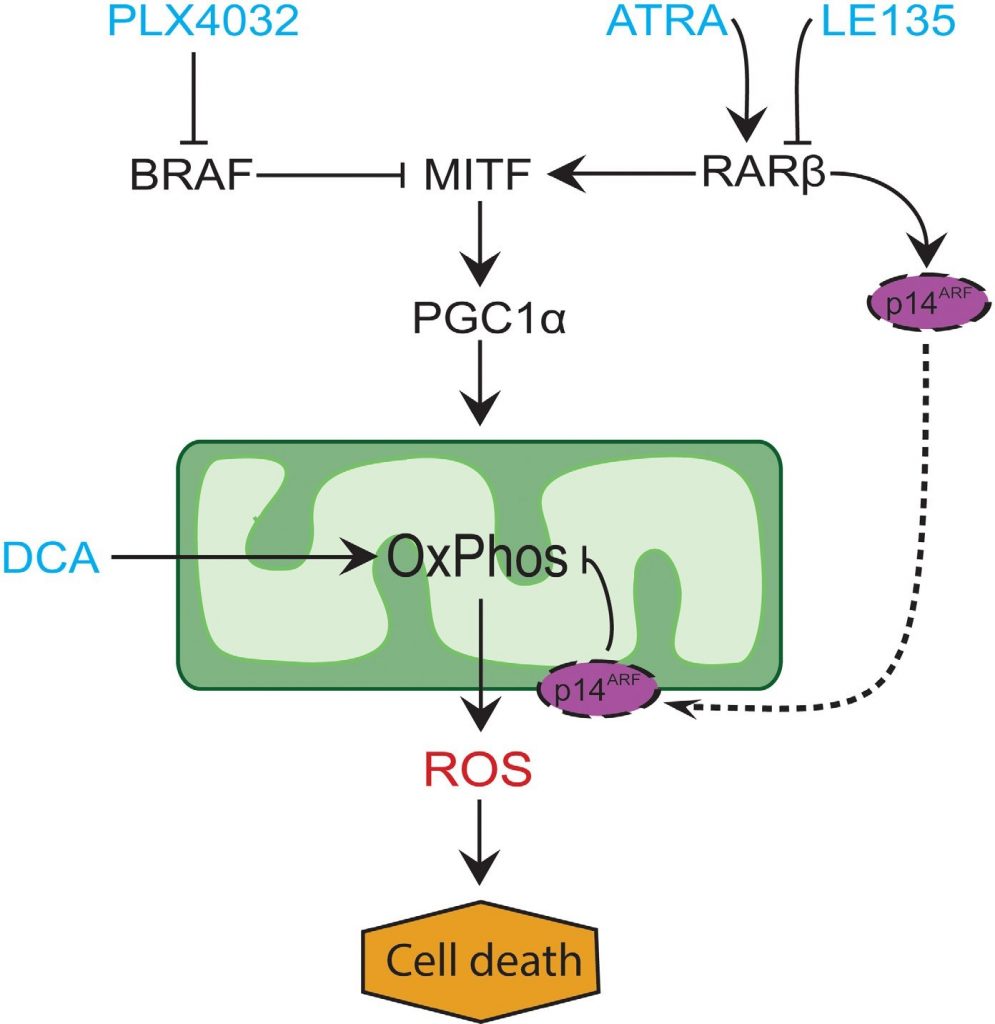
En conclusión, hemos identificado una nueva función de la señalización RARβ en el metabolismo de las células melanocíticas y de melanoma, que podría tener implicaciones clínicas. La capacidad de RARβ para activar la vía MITF-PGC1α, y potencialmente una reducción de la actividad respiratoria mitocondrial dependiente de p14ARF, afecta negativamente a la respuesta terapéutica a la inhibición de BRAF. Sin embargo, el bloqueo de la señalización RARβ promueve la dependencia glucolítica en células de melanoma y potencia el efecto del DCA, lo que potencialmente podría aprovecharse terapéuticamente.
MATERIALES Y MÉTODOS
Reactivos
El dicloroacetato sódico (DCA), la 2-deoxi-D-glucosa (2-DG), el ácido transretinoico total (ATRA) y el LE135 se adquirieron a Sigma-Aldrich. El DCA y la 2-DG se disolvieron en dH2Ohastaalcanzar una concentración de trabajo de 1 M. El ATRA y el LE135 se disolvieron en DMSO hasta alcanzar una concentración de trabajo de 0,1 M. El PLX4032 (vemurafenib) se adquirió a Selleck Chemicals y se disolvió en DMSO hasta alcanzar una concentración de trabajo de 0,05 M.
Cultivo celular
Las líneas celulares de melanoma se obtuvieron de la European Searchable Tumour line Database (ESTDAB, ED) [43]. El estado de estas líneas celulares con respecto a las mutaciones de BRAF y la metilación del promotor de RARB se describió previamente [17]. Los melanocitos epidérmicos humanos primarios (neonatales) procedentes de tejido ligeramente pigmentado (HEMn-LP; denominados melanocitos) se adquirieron a Invitrogen (C0025C). Para los experimentos se utilizaron melanocitos de tres individuos diferentes (lote n.º 200706893 de mar. 2015 y nov. 2016; lote n.º 1583282 de feb. 2017). Las líneas celulares de melanoma se cultivaron a 37 °C bajo un 5 %deCO2 en medio RPMI-1640 suplementado con un 10 % de suero bovino fetal. Las células HEMn-LP se cultivaron en las mismas condiciones en medio 254CF suplementado con un 1% de suplemento de crecimiento de melanocitos humanos (HMGS), incluido forbol 12-miristato 13-acetato (PMA). Para los montajes experimentales, las células HEMn-LP se cultivaron en medio suplementado con HMGS-2 (sin PMA). Todos los medios y suplementos se adquirieron a Invitrogen.
Análisis de viabilidad celular
Seaplicóunensayocon violeta de cristal para evaluar el efecto de los compuestos estudiados sobre la viabilidad celular. Las células se sembraron por duplicado y se trataron con los compuestos correspondientes o con el vehículo de control durante 6 días. El medio y los compuestos de tratamiento se sustituyeron cada 48 h. Los experimentos se repitieron tres veces de forma independiente. Tras el periodo de tratamiento, se eliminó el medio y las células no adheridas, y las células restantes se lavaron con PBS y se fijaron con glutaraldehído durante 15 min. Las células fijadas se incubaron con solución de violeta cristal (0,1% de violeta cristal, 20% de CH3OH) durante 1 h. La cantidad de colorante absorbido por la monocapa, proporcional al número de células viables adheridas al fondo del pocillo, se cuantificó extrayendo el color con ácido acético al 10% y midiendo la absorbancia a una longitud de onda de 595 nm. La viabilidad relativa tras el tratamiento con ATRA, LE135 o PLX4032 se utilizó para determinar la concentración inhibitoria semimáxima (IC50). Trazando la curva dosis-respuesta, el valor IC50 se estimó como la concentración en el punto del 50% de viabilidad celular.
Purificación de ADN y ARN
El ADN para la cuantificación del ADNmt y el ARN para la síntesis de ADNc se purificaron simultáneamente con el minikit AllPrep DNA/RNA/Protein (Qiagen) siguiendo el protocolo proporcionado.
Análisis de expresión
La síntesis de ADNc se realizó con el qScript™ XLT cDNA SuperMix (Quanta Bioscience). La expresión génica de PGC1α, MITF, RARβ, p14ARF, UCP2, ATP5g1, COX5A y NDUFS3 se determinó con PCR cuantitativa en tiempo real en el LightCycler 2.0 de Roche utilizando el kit LightCycler FastStart DNA MasterPLUS SYBR Green I (Roche). Los cebadores se enumeran en la Tabla suplementaria 1.
| Gen | Cebador directo | Cebador inverso |
| PGC1α* | GTAAATCTGCGGGATGATGG | AATTGCTTGCGTCCACAAA |
| MITF** | CCGTCTCTCACTGGATTGGT | TACTTGGTGGTTTTCGAG |
| p14ARF*** | CCCTCGTGCTGATGCTACTGA | CATGACCTGGTCTTCTAGGAAGC |
| RARβ*** | TCCTGGATTTCTACACTGCG | AAGCAGGGTTTGTACACTCG |
| UCP2 | AAGACCATTGCCCGAGAGG | TTGGCTTTCAGGAGGGCAT |
| ATP5g1* | ATCATTGGCTATGCCAGGAA | ATGGCGAAGGATGAGGA |
| COX5A | GGGAATTGCGTAAAGGGATAA | TCCTGCTTTGTCCTTAACAACC |
| NDUFS3* | GCTGACGCCCATTGAGTCTG | GGAACTCTTGGGCCAACTCC |
| RPLP0 | ACTAAAATCTCCAGGGGCACC | ATGACCAGCCCAAAGGAGAA |
*Secuencias de cebadores publicadas por Vázquez et al. 2013 [22].
**Secuencias de cebadores publicadas por Haq et al. 2014 [11].
***Secuencias de cebadores publicadas por Dahl et al. 2013 [17].
Immunoblotting
Las muestras se prepararon a partir de frascos de cultivo celular con tampón de lisis (SLB) suplementado con β-mercaptoetanol incoloro (BPB), Phospho-Stop e inhibidor de proteasas (Thermo Fisher Scientific). Los lisados celulares se aclararon mediante centrifugación a 20.000 rpm durante 3 min. La concentración de proteínas se midió utilizando el kit Qubit Protein Assay (Thermo Fisher Scientific), y 50 μg de proteína de cada muestra se cargaron en un gel de 10 pocillos SDS, 4-12% Bis-Tris NuPage (Invitrogen). A continuación, las proteínas se separaron a 80 V durante 30 min, seguidos de 110 V hasta su finalización. El blotting se realizó con una unidad de transferencia semiseca sobre una membrana de nitrocelulosa ECL a 3,3 mA/1 cm2/1 h/gel. A continuación, la membrana se tiñó con Ponceau. La membrana se bloqueó en leche al 5% durante 1 h, después se lavó dos veces durante 5 min con TBST y se tiñó con anticuerpos anti-AMPK o anti-p-AMPK (Thr172) (Cell Signaling; 1:2000) en BSA al 5% a 4°C y con un anticuerpo anti ciclofilina A (Cell Signaling; 1:5000) como control de carga. Después de tres ciclos de 10 minutos de lavado con TBST, la membrana se tiñó con el anticuerpo secundario (anti-conejo; DakoCytomation; 1:2000) durante 1 h a temperatura ambiente, seguido de otros 3 ciclos de lavado. Las proteínas se visualizaron con ECL Plus Western Blotting Substrate (Thermo Fisher Scientific) 1:1 durante 2-3 min.
Análisis metabólico
El análisis metabólico se realizó en líneas celulares de melanoma y melanocitos utilizando un analizador Seahorse XFe96 (Seahorse Bioscience, Billerica, MA), que realiza mediciones en tiempo real de la tasa de acidificación extracelular (ECAR) y la tasa de consumo de oxígeno (OCR). Se sembraron 20.000 células por pocillo en microplacas de cultivo celular Seahorse 24 h antes de realizar las mediciones. Los cambios en la actividad basal y la capacidad de los sistemas energéticos mitocondrial y glucolítico se determinaron utilizando el Mito Stress Test Kit y el Glycolysis Stress Test Kit (Agilent Technologies). Los ensayos se realizaron de acuerdo con los protocolos proporcionados. La prueba de estrés mitótico se realizó en el medio de cultivo habitual, mientras que en la prueba de estrés glucolítico, el medio se sustituyó por medio Seahorse XF Base suplementado con L-glutamina (2 mM), pH ajustado a 7,4, 1 h antes de las mediciones. Para exposiciones más prolongadas (>24 h), las células se trataron en frascos de cultivo antes de la siembra. Todos los resultados se normalizaron con respecto al número de células sembradas, ya que las concentraciones de ATRA y LE135 utilizadas no afectaron al crecimiento celular durante 24 h. El protocolo para realizar los ensayos en la máquina Seahorse incluía ciclos de 3 min de mezcla/3 min de medición. Se realizaron tres experimentos independientes con 6 réplicas de cada muestra.
Transfección
La línea celular de melanoma ED-007 se transfectó con vectores de expresión pEGFP (control) o pEGFP-p14ARF (2 μg de vector/2×106 células), ambos con GFP como gen informador. Los constructos se obtuvieron como se describió previamente [19]. La transfección se realizó utilizando la tecnología de nucleofección Amaxa, tampón V, programa T-020, siguiendo el protocolo recomendado por el fabricante. El éxito de la transfección se verificó visualmente. Los clones estables se seleccionaron utilizando 400 μg/ml de G418 (geneticina; Thermo Fisher Scientific). En el montaje experimental, las células se sembraron sin G418.
Cuantificación del ADN mitocondrial
ElADNmitocondrialsecuantificó mediante reacción en cadena de la polimerasa digital en gotas (ddPCR) utilizando el sistema QX200 (BioRad Laboratories, Hercules, CA, EE.UU.). Se utilizaron aproximadamente 0,5 ng de ADN para cada reacción. El número de copias mitocondrial se determinó calculando la relación entre un sitio de ADN mitocondrial (mtMinArc) y un locus nuclear de una sola copia (β2m), tal y como describen Phillips et al. [44]. Los cebadores, sondas y condiciones experimentales se enumeran en la Tabla suplementaria 2.
| mtMinArc | β2m | |
| Cebador directo | CTAAATAGCCCACACGTTCCC | GCTGGGTAGCTCTAAACAATGTATTCA |
| Cebador inverso | AGAGCTCCCGTGAGTGGTTA | CCATGTACTAACAAATGTCTAAAATGGT |
| Sonda | 6FAM-CATCACGATGGATCACAGGT(NFQ) | VIC-CAGCAGCCTATTCTGC(NFQ) |
| Primer conc. | 75 nM | 500 nM |
| Temp. de recocido | 50°C | 52°C |
| Nº de ciclos | 40 | 40 |
*Secuencias de primers y sondas publicadas por Phillips et al. [44].
Análisis estadístico
Las diferencias entre conjuntos de datos independientes se determinaron con la prueba t de Student. Para el análisis estadístico de la varianza entre los distintos tratamientos se utilizó el ANOVA de muestras emparejadas de una vía. Para determinar la significación estadística se utilizó la prueba multicomparativa de diferencia significativa honesta (HSD) de Tukey.
Contribución de los autores
CA y PG planificaron y organizaron el estudio. CA realizó la mayoría de los experimentos y el procesamiento de los datos, incluyendo el cultivo celular, los protocolos de tratamiento, el análisis metabólico y las estadísticas. CD planificó y realizó la transfección EGFP-p14ARF y las mediciones de ADNmt y ayudó a interpretar los resultados. AA realizó y optimizó los protocolos de inmunotransferencia y PCR cuantitativa. AC realizó ensayos de cultivo celular. CA y PG escribió el manuscrito con las contribuciones y las ediciones de CD, AA y AC. El manuscrito final fue leído y aprobado por todos los autores.
CONFLICTOS DE INTERESES
Los autores declaran no tener ningún conflicto de intereses.
FINANCIACIÓN
Este estudio ha sido financiado por la Sociedad Danesa del Cáncer
REFERENCIAS
1 Tasas de supervivencia del cáncer de piel melanoma, por estadios. (cancer.org: Sociedad Americana del Cáncer).2 Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, Chodon T, Nelson SF, McArthur G, et al. Melanomas acquire resistance to B-RAF (V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010; 468: 973-7. https://doi.org/10.1038/nature09626.
3 Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP, Barretina J, Caponigro G, Hieronymus H, Murray RR, et al. COT impulsa la resistencia a la inhibición de RAF a través de la reactivación de la vía MAP quinasa. Nature. 2010; 468: 968-72. https://doi.org/10.1038/nature09627.
4 Miller AJ, Mihm MC Jr. Melanoma. N Engl J Med. 2006; 355: 51-65. https://doi.org/10.1056/NEJMra052166.
5 Abildgaard C, Guldberg P. Molecular drivers of cellular metabolic reprogramming in melanoma. Trends Mol Med. 2015; 21: 164-71. https://doi.org/10.1016/j.molmed.2014.12.007.
6 Ratnikov BI, Scott DA, Osterman AL, Smith JW, Ronai ZA. Recableado metabólico en el melanoma. Oncogene. 2017; 36: 147-57. https://doi.org/10.1038/onc.2016.198.
7 Warburg O, Wind F, Negelein E. El metabolismo de los tumores en el cuerpo. J Gen Physiol. 1927; 8: 519-30.
8 Hall A, Meyle KD, Lange MK, Klima M, Sanderhoff M, Dahl C, Abildgaard C, Thorup K, Moghimi SM, Jensen PB, Bartek J, Guldberg P, Christensen C. Dysfunctional oxidative phosphorylation makes malignant melanoma cells addicted to glycolysis driven by the (V600E) BRAF oncogene. Oncotarget. 2013; 4: 584-99. https://doi.org/10.18632/oncotarget.965.
9 Parmenter TJ, Kleinschmidt M, Kinross KM, Bond ST, Li J, Kaadige MR, Rao A, Sheppard KE, Hugo W, Pupo GM, Pearson RB, McGee SL, Long GV, et al. Response of BRAF-mutant melanoma to BRAF inhibition is mediated by a network of transcriptional regulators of glycolysis. Cáncer Discov. 2014; 4: 423-33. https://doi.org/10.1158/2159-8290.CD-13-0440.
10 McArthur GA, Puzanov I, Amaravadi R, Ribas A, Chapman P, Kim KB, Sosman JA, Lee RJ, Nolop K, Flaherty KT, Callahan J, Hicks RJ. Marked, homogeneous, and early [18F]fluorodeoxyglucose-positron emission tomography responses to vemurafenib in BRAF-mutant advanced melanoma. J Clin Oncol. 2012; 30: 1628-34. https://doi.org/10.1200/JCO.2011.39.1938.
11 Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, Frederick DT, Hurley AD, Nellore A, Kung AL, Wargo JA, Song JS, Fisher DE, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell. 2013; 23: 302-15. https://doi.org/10.1016/j.ccr.2013.02.003.
12 Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011; 364: 2507-16. https://doi.org/10.1056/NEJMoa1103782.
13 Corazao-Rozas P, Guerreschi P, Jendoubi M, Andre F, Jonneaux A, Scalbert C, Garcon G, Malet-Martino M, Balayssac S, Rocchi S, Savina A, Formstecher P, Mortier L, et al. Mitochondrial oxidative stress is the Achille’s heel of melanoma cells resistant to Braf-mutant inhibitor. Oncotarget. 2013; 4: 1986-98. https://doi.org/10.18632/oncotarget.1420.
14 Zhang G, Frederick DT, Wu L, Wei Z, Krepler C, Srinivasan S, Chae YC, Xu X, Choi H, Dimwamwa E, Ope O, Shannan B, Basu D, et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J Clin Invest. 2016; 126: 1834-56. https://doi.org/10.1172/JCI82661.
15 Livingstone E, Swann S, Lilla C, Schadendorf D, Roesch A. Combining BRAF(V) (600E) inhibition with modulators of the mitochondrial bioenergy metabolism to overcome drug resistance in metastatic melanoma. Exp Dermatol. 2015; 24: 709-10. https://doi.org/10.1111/exd.12718.
16 Hoon DS, Spugnardi M, Kuo C, Huang SK, Morton DL, Taback B. Profiling epigenetic inactivation of tumor suppressor genes in tumors and plasma from cutaneous melanoma patients. Oncogene. 2004; 23: 4014-22. https://doi.org/10.1038/sj.onc.1207505.
17 Dahl C, Christensen C, Jonsson G, Lorentzen A, Skjodt ML, Borg A, Pawelec G, Guldberg P. Mutual exclusivity analysis of genetic and epigenetic drivers in melanoma identifies a link between p14 ARF and RARbeta signaling. Mol Cancer Res. 2013; 11: 1166-78. https://doi.org/10.1158/1541-7786.MCR-13-0006.
18 Lotan R, Lotan D. Enhancement of melanotic expression in cultured mouse melanoma cells by retinoids. J Cell Physiol. 1981; 106: 179-89. https://doi.org/10.1002/jcp.1041060203.
19 Christensen C, Bartkova J, Mistrik M, Hall A, Lange MK, Ralfkiaer U, Bartek J, Guldberg P. A short acidic motif in ARF guards against mitochondrial dysfunction and melanoma susceptibility. Nat Commun. 2014; 5: 5348. https://doi.org/10.1038/ncomms6348.
20 Baldea I, Costin GE, Shellman Y, Kechris K, Olteanu ED, Filip A, Cosgarea MR, Norris DA, Birlea SA. Biphasic pro-melanogenic and pro-apoptotic effects of all-trans-retinoic acid (ATRA) on human melanocytes: time-course study. J Dermatol Sci. 2013; 72: 168-76. https://doi.org/10.1016/j.jdermsci.2013.06.004.
21 Kawakami T, Ohgushi A, Hirobe T, Soma Y. Analysis of the effects of all-trans retinoic acid on human melanocytes and melanoblasts in vitro. J Dermatol. 2017; 44: 93-4. https://doi.org/10.1111/1346-8138.13477.
22 Vázquez F, Lim JH, Chim H, Bhalla K, Girnun G, Pierce K, Clish CB, Granter SR, Widlund HR, Spiegelman BM, Puigserver P. La expresión de PGC1alfa define un subconjunto de tumores de melanoma humano con mayor capacidad mitocondrial y resistencia al estrés oxidativo. Cancer Cell. 2013; 23: 287-301. https://doi.org/10.1016/j.ccr.2012.11.020.
23 Donadelli M, Dando I, Fiorini C, Palmieri M. UCP2, una proteína mitocondrial regulada a múltiples niveles. Cell Mol Life Sci. 2014; 71: 1171-90. https://doi.org/10.1007/s00018-013-1407-0.
24 Oberkofler H, Klein K, Felder TK, Krempler F, Patsch W. Role of peroxisome proliferator-activated receptor-gamma coactivator-1alpha in the transcriptional regulation of the human uncoupling protein 2 gene in INS-1E cells. Endocrinology. 2006; 147: 966-76. https://doi.org/10.1210/en.2005-0817.
25 Arigony AL, de Oliveira IM, Machado M, Bordin DL, Bergter L, Pra D, Henriques JA. La influencia de los micronutrientes en el cultivo celular: una reflexión sobre la viabilidad y la estabilidad genómica. Biomed Res Int. 2013; 2013: 597282. https://doi.org/10.1155/2013/597282.
26 Li Y, Hashimoto Y, Agadir A, Kagechika H, Zhang X. Identification of a novel class of retinoic acid receptor beta-selective retinoid antagonists and their inhibitory effects on AP-1 activity and retinoic acid-induced apoptosis in human breast cancer cells. J Biol Chem. 1999; 274: 15360-6.
27 de The H, Marchio A, Tiollais P, Dejean A. Differential expression and ligand regulation of the retinoic acid receptor alpha and beta genes. EMBO J. 1989; 8: 429-33.
28 Hardie DG, Hawley SA. AMP-activated protein kinase: the energy charge hypothesis revisited. Bioessays. 2001; 23: 1112-9. https://doi.org/10.1002/bies.10009.
29 potencia su respuesta a la inhibición de BRAFV600E. J Transl Med. 2014; 12: 247. https://doi.org/10.1186/s12967-014-0247-5.
30 Populo H, Caldas R, Lopes JM, Pardal J, Maximo V, Soares P. Overexpression of pyruvate dehydrogenase kinase supports dichloroacetate as a candidate for cutaneous melanoma therapy. Expert Opin Ther Targets. 2015; 19: 733-45. https://doi.org/10.1517/14728222.2015.1045416.
31 Kluza J, Corazao-Rozas P, Touil Y, Jendoubi M, Maire C, Guerreschi P, Jonneaux A, Ballot C, Balayssac S, Valable S, Corroyer-Dulmont A, Bernaudin M, Malet-Martino M, et al. Inactivation of the HIF-1alpha/PDK3 signaling axis drives melanoma towards mitochondrial oxidative metabolism and potentiates the therapeutic activity of pro-oxidants. Cancer Res. 2012; 72: 5035-47. https://doi.org/10.1158/0008-5472.CAN-12-0979.
32 Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, van der Burg SH, Verdegaal EM, Cascante M, Shlomi T, Gottlieb E, Peeper DS. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature. 2013; 498: 109-12. https://doi.org/10.1038/nature12154.
33 Murholm M, Isidor MS, Basse AL, Winther S, Sorensen C, Skovgaard-Petersen J, Nielsen MM, Hansen AS, Quistorff B, Hansen JB. El ácido retinoico tiene diferentes efectos sobre la expresión de UCP1 en adipocitos de ratón y humanos. BMC Cell Biol. 2013; 14: 41. https://doi.org/10.1186/1471-2121-14-41.
34 Jin W, Xu YP, Yang AH, Xing YQ. In vitro induction and differentiation of umbilical cord mesenchymal stem cells into neuron-like cells by all-trans retinoic acid. Int J Ophthalmol. 2015; 8: 250-6. https://doi.org/10.3980/j.issn.2222-3959.2015.02.07.
35 Tourniaire F, Musinovic H, Gouranton E, Astier J, Marcotorchino J, Arreguin A, Bernot D, Palou A, Bonet ML, Ribot J, Landrier JF. All-trans retinoic acid induces oxidative phosphorylation and mitochondria biogenesis in adipocytes. J Lipid Res. 2015; 56: 1100-9. https://doi.org/10.1194/jlr.M053652.
36 Tripathy S, Chapman JD, Han CY, Hogarth CA, Arnold SL, Onken J, Kent T, Goodlett DR, Isoherranen N. All-trans-retinoic acid enhances mitochondrial function in models of human liver. Mol Pharmacol. 2016; 89: 560-74. https://doi.org/10.1124/mol.116.103697.
37 Watabe H, Soma Y, Ito M, Kawa Y, Mizoguchi M. All-trans retinoic acid induces differentiation and apoptosis of murine melanocyte precursors with induction of the microphthalmia-associated transcription factor. J Invest Dermatol. 2002; 118: 35-42. https://doi.org/10.1046/j.0022-202x.2001.01614.x.
38 De Preter G, Neveu MA, Danhier P, Brisson L, Payen VL, Porporato PE, Jordan BF, Sonveaux P, Gallez B. Inhibition of the pentose phosphate pathway by dichloroacetate unravels a missing link between aerobic glycolysis and cancer cell proliferation. Oncotarget. 2016; 7: 2910-20. https://doi.org/10.18632/oncotarget.6272.
39 Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer. 2008; 99: 989-94. https://doi.org/10.1038/sj.bjc.6604554.
40 Corazao-Rozas P, Guerreschi P, Andre F, Gabert PE, Lancel S, Dekiouk S, Fontaine D, Tardivel M, Savina A, Quesnel B, Mortier L, Marchetti P, Kluza J. Mitochondrial oxidative phosphorylation controls cancer cell’s life and death decisions upon exposure to MAPK inhibitors. Oncotarget. 2016; 7: 39473-85. https://doi.org/10.18632/oncotarget.7790.
41 Bauer D, Werth F, Nguyen HA, Kiecker F, Eberle J. Critical role of reactive oxygen species (ROS) for synergistic enhancement of apoptosis by vemurafenib and the potassium channel inhibitor TRAM-34 in melanoma cells. Cell Death Dis. 2017; 8: e2594. https://doi.org/10.1038/cddis.2017.6.
42 Chen MC, Hsu SL, Lin H, Yang TY. Ácido retinoico y tratamiento del cáncer. Biomedicina (Taipéi). 2014; 4: 22. https://doi.org/10.7603/s40681-014-0022-1.
43 Robinson J, Roberts CH, Dodi IA, Madrigal JA, Pawelec G, Wedel L, Marsh SG. The European searchable tumour line database. Cancer Immunol Immunother. 2009; 58: 1501-6. https://doi.org/10.1007/s00262-008-0656-5.
44 Phillips NR, Sprouse ML, Roby RK. Simultaneous quantification of mitochondrial DNA copy number and deletion ratio: a multiplex real-time PCR assay. Sci Rep. 2014; 4: 3887. https://doi.org/10.1038/srep03887.