Ronen Shavit1, Maya Ilouze, Tali Feinberg, Yaacov Richard Lawrence, Yossi Tzur, Nir Peled
1 Centro de Investigación y Detección del Cáncer Torácico, Centro Médico Sheba Tel Hashomer, Ramat-Gan, 52621, POB 244, Israel.
correo electrónico: [email protected]
URL: http://medicine.mytau.org/peled/
Y. R.Lawrence
Center for Translational Research in Radiation Oncology, Sheba Medical Center, Ramat-Gan, Israel
M. Ilouze : N. Peled
Thoracic Cancer Service, Davidoff Cancer Center, Rabin Medical Center, Petach Tikva, Israel
Correspondencia: [email protected]
Aceptado: 10 de diciembre de 2014
Publicado: 7 de enero de 2015
Resumen
Introducción: El cáncer de pulmón es la principal causa de muerte por cáncer. La radioterapia desempeña un papel clave en su tratamiento. La radiación ionizante induce la muerte celular a través de aberraciones cromosómicas, que desencadenan la catástrofe mitótica y la apoptosis. Sin embargo, muchos pacientes con cáncer de pulmón muestran resistencia a la radiación. El dicloroacetato (DCA) es una pequeña molécula que puede promover la activación mitocondrial mediante el aumento de la afluencia de piruvato. Aquí, probamos si el DCA puede aumentar la sensibilidad de las células de cáncer de pulmón no microcítico (CPNM) a la radiación a través de este mecanismo.
Métodos: Se analizó la sensibilidad de dos líneas celulares representativas de CPNM (A549 y H1299) a la radiación con y sin preexposición al DCA. La eficacia del tratamiento se evaluó mediante un ensayo de supervivencia clonogénica. Se utilizó un analizador de flujo extracelular para evaluar el efecto del DCA en el consumo de oxígeno celular como marcador sustitutivo de la actividad mitocondrial.
Resultados: Se observó que el DCA aumenta la tasa de consumo de oxígeno en las células A549 y H1299 en un 60% (p = 0,0037) y un 20% (p = 0,0039), respectivamente. La preexposición al DCA una hora antes de la radiación aumentó la tasa de muerte citotóxica 4 veces en las células A549 (55 a 13%, p = 0,004) y 2 veces en las células H1299 (35 a 17%, p = 0,28) respectivamente, en comparación con la radiación sola.
Conclusiones: La inducción mitocondrial por el DCA puede servir como radiosensibilizador en el cáncer de pulmón no microcítico.
Palabras clave: CPNM; DCA; Mitocondrias; Radiación; Radiosensibilizador; Efecto Warburg
sociedad Internacional de Oncología Celular 2015
INTRODUCCIÓN
El cáncer de pulmón es la principal causa de muerte por cáncer en Estados Unidos, con una tasa de supervivencia global a 5 años para todos los estadios de ~17 % [1-4]. La radioterapia (RT) desempeña un papel importante en el manejo clínico de los pacientes con cáncer de pulmón, particularmente en aquellos con enfermedad en estadio IIIB que son candidatos a quimio-radioterapia definitiva. Además, la RT puede aplicarse como terapia neoadyuvante o adyuvante en el estadio IIIA, o de forma ablativa cuando se aplica radioterapia corporal estereotáctica (SBRT). La neumonitis inducida por la radiación (NIR) es el factor limitante cuando se trata a pacientes con RT. Para minimizar la RIP, los oncólogos intentan mantener el V20 (es decir, el porcentaje del volumen pulmonar que recibe una dosis de radiación de ≥20 Gy) por debajo del 22 % [5]. Los radiosensibilizadores pueden aumentar la eficacia citotóxica de la radiación, mejorando así potencialmente las tasas de curación sin aumentar el V20.
La RT mata las células provocando roturas de la doble cadena del ADN. Las roturas de ADN no reparadas dan lugar a aberraciones cromosómicas que, a su vez, conducen a la «catástrofe mitótica», un modo de muerte celular que resulta de la entrada prematura o inapropiada de las células en mitosis [6,7]. Además, la radiación puede afectar directamente a las membranas y orgánulos celulares. Aunque estos últimos cambios aún no se conocen bien, pueden provocar cambios en la transducción de señales, la expresión génica, la estabilidad de las proteínas, los estados redox celulares y la regulación del ciclo celular, todo lo cual puede conducir a la apoptosis [8]. Además, la radiación puede inducir la producción de especies reactivas de oxígeno (ROS) mitocondriales, acompañada de una regulación al alza de las funciones de la cadena de transporte de electrones mitocondrial, aumentando así el potencial de membrana mitocondrial, la respiración mitocondrial y la producción de ATP mitocondrial [9,10]. Sin embargo, las alteraciones genéticas limitan la capacidad de las células cancerosas para someterse a la apoptosis, lo que sugiere que los microambientes tumorales hipóxicos pueden ejercer una presión selectiva hacia un fenotipo canceroso resistente a la apoptosis y, como resultado, resistente a la RT [11]. Debido a este fenotipo resistente a la RT y a los importantes efectos secundarios causados por la propia RT, es de vital importancia explorar formas de radiosensibilizar las células de cáncer de pulmón para disminuir las dosis de radiación y mejorar las respuestas a la terapia.
Muchas células tumorales presentan niveles elevados de captación de glucosa y niveles reducidos de fosforilación oxidativa. Esta paradójica hiperglicólisis y falta de actividad mitocondrial en presencia de oxígeno se conoce como efecto Warburg (representado en la Fig. 1) [12]. El metabolismo único de la mayoría de los tumores sólidos se deriva de la remodelación de las funciones mitocondriales para producir un fenotipo glucolítico y una fuerte resistencia a la apoptosis [13]. Cada vez hay más pruebas que indican que las mitocondrias pueden ser las dianas principales de los tratamientos contra el cáncer [14-17]. La remodelación específica del cáncer puede revertirse mediante una pequeña molécula llamada dicloroacetato (DCA) [18], que inhibe la piruvato deshidrogenasa cinasa (PDK), aumentando así la afluencia de piruvato a la mitocondria y promoviendo su oxidación a través de la actividad mitocondrial por encima de la glucólisis en varios tipos de cáncer (representado en la Fig. 1) [13,18]. Un aumento de la actividad mitocondrial provoca un aumento tanto de la cantidad como de la extensión de la liberación de radicales libres (ROS) en la célula, lo que conduce a un aumento de la actividad apoptótica celular [13].
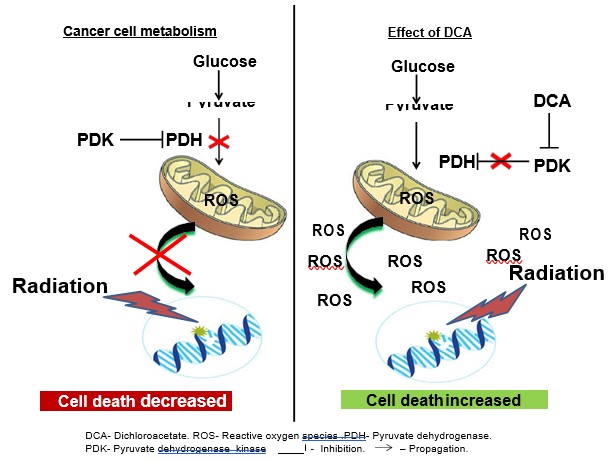
En este trabajo investigamos el posible efecto radiosensibilizador del DCA en células de CPNM. Nuestros resultados indican que la activación mitocondrial por DCA aumenta la sensibilidad a la radiación tanto en células derivadas de NSCLC A549 como H1299.
Materiales y métodos
Cultivos celulares
En este estudio se utilizaron dos líneas celulares de cáncer de pulmón no microcítico (CPNM): A549, derivada de un adenocarcinoma humano, y H1299, derivada de un carcinoma humano de células grandes. Ambas líneas celulares se adquirieron al ATCC. Las células se cultivaron en medio RPMI-1640 suplementado con un 10 % de suero bovino fetal (FBS), un 1 % de penicilina/estreptomicina y un 1 % de glutamato a 37 °C en una atmósfera humidificadacon un 5 % deCO2.
Condiciones de tratamiento
Se añadiódicloroacetato(DCA, Sigma 34795) a los medios de cultivo durante 1 h antes de la radiación a 10 y 20 mM en el caso de las células H1299 y a 40 y 60 mM en el caso de las células A549, de acuerdo con los respectivos valores IC50. Las células se irradiaron con un irradiador Kimtron Polaris a una tasa de dosis de 0,9 Gy/min a temperatura ambiente.
Ensayo de supervivencia clonogénica
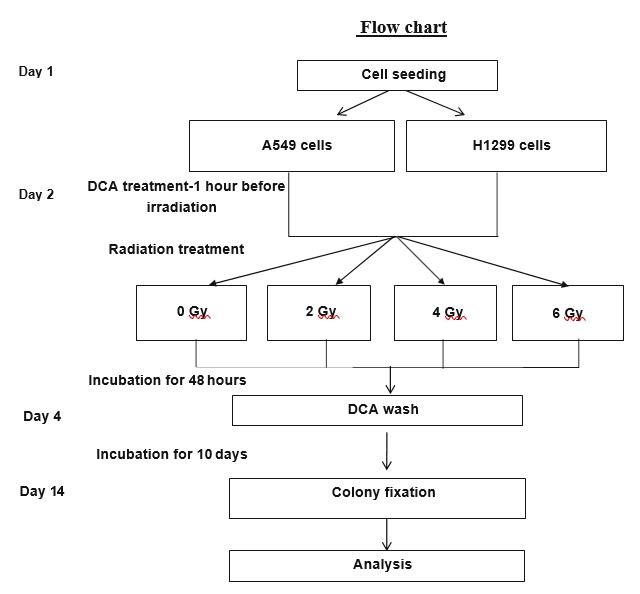
El ensayo de supervivencia clonogénica que utilizamos ha sido descrito previamente [19]. Brevemente, las células se desprendieron con tripsina, se suspendieron en medio de cultivo completo, se contaron y se sembraron en placas de 3 ml a una densidad de 200 células por placa, y luego se dejaron adherir y estabilizar durante la noche. Al día siguiente, los grupos de prueba se trataron con DCA (o control) 1 h antes de la radiación, a las concentraciones indicadas. Las células se irradiaron con dosis de 0, 2, 4 y 6 Gy. Tras 48 h de incubación, se retiraron los medios de cultivo y se sustituyeron por medios de cultivo frescos sin DCA (todos los grupos). Las células se incubaron durante otros 10 días para permitir la formación de colonias. A continuación, se fijaron las colonias y se tiñeron con violeta cristal al 0,5% en etanol al 96%. Se contaron las colonias que contenían al menos 50 células viables (Fig. 2). Cada experimento se realizó por duplicado y la media y la desviación estándar se calcularon a partir de 3 experimentos independientes.
Evaluación de la sinergia
Para evaluar si los esquemas de tratamiento muestran efectos sinérgicos, se realizaron gráficos de medias con desviaciones estándar de los porcentajes de supervivencia relativos a las dosis de radiación. Se calcularon los porcentajes de supervivencia, tanto de los grupos tratados como de los no tratados con DCA, en relación con los de las células no irradiadas:
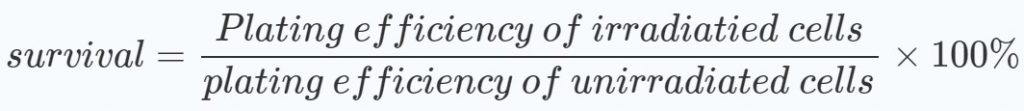
donde:
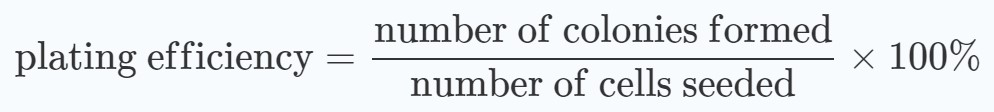
Específicamente, la formación de colonias de los grupos tratados con DCA y no tratados con DCA sin tratamiento de radiación se normalizaron al 100 %. Los efectos sinérgicos se determinaron mediante un ANOVA de dos vías (valor p <0,05) [20].
Ensayo de consumo de oxígeno
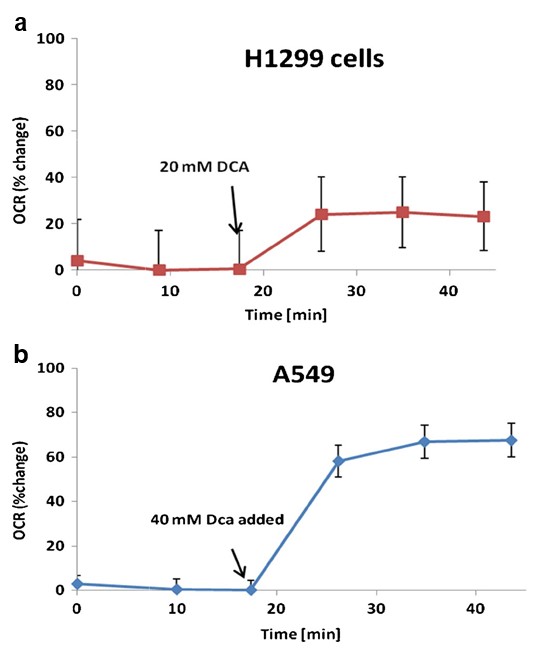
El consumo de oxígeno se utilizó como marcador sustitutivo de la actividad mitocondrial. Las mediciones se llevaron a cabo utilizando un analizador de flujo extracelular XF24 (Seahorse Biosciences). Este dispositivo utiliza sensores ópticos basados en fluorescencia y placas multipocillo a medida para realizar mediciones repetidas del consumo de oxígeno de células intactas que crecen en monocapa. Las células H1299 y A549 se sembraron en placas de cultivo celular Seahorse XF24 a 10.000 y 20.000 células por pocillo, respectivamente, en medio de crecimiento. Las células se incubaron durante 48 h a 37 °C en un 5 % deCO2. A continuación, las células se lavaron y transfirieron a medio de ensayo XF/PBS, y se incubaron durante 60 min a 37 °C sinCO2 antes de iniciar el experimento. Tras establecer las tasas de consumo de oxígeno de referencia, se añadieron 20 y 40 mM de DCA a las células H1299 y A549, respectivamente. Las mediciones del consumo de oxígeno continuaron durante 25 minutos. Se obtuvieron datos de al menos tres placas replicadas por línea celular. Los resultados se representan como el porcentaje de la tasa de respiración basal.
Análisis estadístico
La significación estadística se evaluó mediante la prueba t
Student. La sinergia se evaluó mediante un ANOVA de dos vías [20]. Un valor de p< 0,05 se consideró significativo.
Resultados
ElDCA radiosensibiliza las células de CPNM
Se sabe queel dicloroacetato(DCA) aumenta la actividad mitocondrial [10,15] y anteriormente se ha demostrado que actúa como radiosensibilizador en varias líneas celulares de cáncer derivadas de tumores de próstata, colorrectales y cerebrales [21, 22]. Aquí se investigó su efecto en dos líneas celulares derivadas de cáncer de pulmón no microcítico (CPNM), a saber, A549 (adenocarcinoma) y H1299 (carcinoma de células grandes). Las concentraciones de DCA utilizadas para cada línea celular se ajustaron a los respectivos valores IC50 (A549: 63 mM y H1299: 36 mM; Fig. 3a y b).
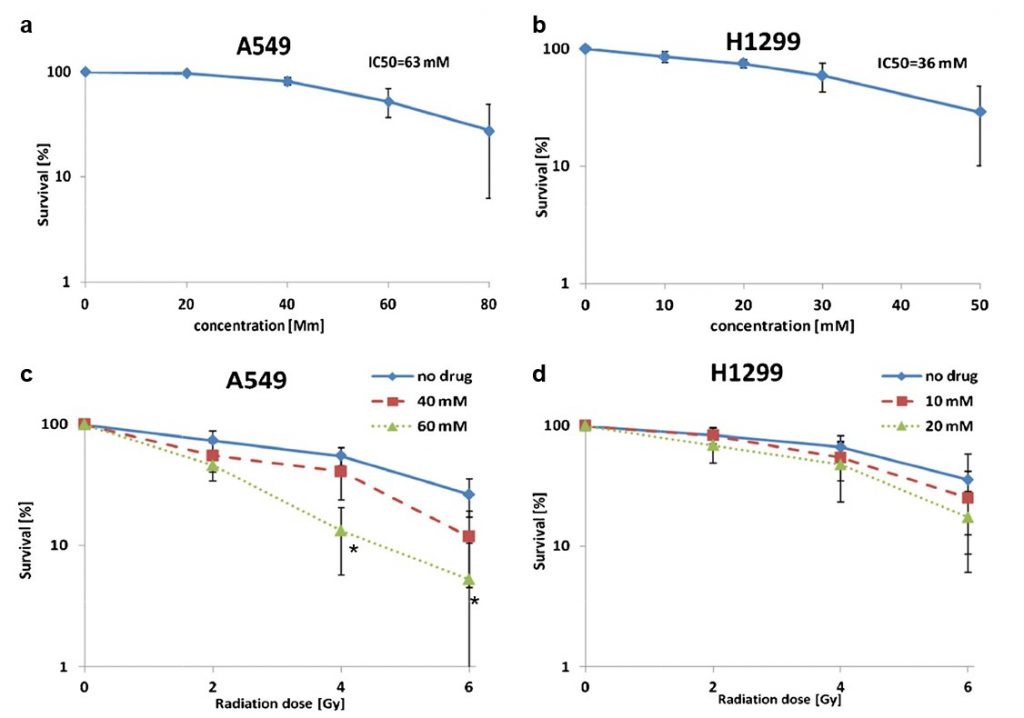
Descubrimos que la exposición previa a la radiación con DCA aumentaba la muerte celular tanto en muestras de células A549 como H1299. El efecto máximo en las células A549 se observó a 4 Gy, donde el tratamiento con DCA (40 y 60 mM) disminuyó la supervivencia celular de ~55 a 40 % (p= 0,27) y 13 % (p= 0,004), respectivamente (Fig. 3c). En las células H1299, el efecto máximo se alcanzó a los 6 Gy. A esta dosis de radiación, el tratamiento con DCA (10 y 20 mM) disminuyó la supervivencia celular del 35 % (sólo radiación) al 25 y 17 %, respectivamente (no significativo estadísticamente; Fig. 3d). La exposición previa de las células A549 al DCA (60 mM) multiplicó por 4 el efecto citotóxico de la radiación de 4 Gy en comparación con la radiación sola. Un análisis ANOVA de dos vías indicó un efecto sinérgico sólo en las células A549.
El DCAaumenta el consumo de oxígeno en las células A549 y H1299
Una mayor tasa de consumo de oxígeno celular (OCR) es una indicación de una mayor actividad mitocondrial [23]. Hemos observado que el DCA aumenta el OCR tanto en las células A549 como en las H1299 en un 60% (p= 0,0037) y un 20% (p= 0,0039), respectivamente, en comparación con el OCR basal (Fig. 4).
Discusión
Al comprobar la sensibilidad a la radiación con y sin preexposición a dicloroacetato (DCA), descubrimos que la activación mitocondrial por DCA puede radiosensibilizar dos líneas celulares distintas derivadas de cáncer de pulmón de células no pequeñas (CPCNP) (es decir, A549 y H1299). En este estudio de prueba de concepto, se utilizó una amplia gama de concentraciones de DCA, con el objetivo de adaptar la dosis al valor IC50 de cada célula. El efecto sinérgico del DCA fue estadísticamente significativo en las células A549, mientras que sólo se aproximó a la significación en las células H1299. Posiblemente, puede obtenerse un efecto aún más robusto con dosis más altas de DCA. También demostramos que el DCA aumenta el consumo de oxígeno tanto en las células A549 como en las H1299, que es un marcador sustituto del aumento de la actividad mitocondrial.
La radiación ionizante regula al alza la función de la cadena de transporte de electrones mitocondrial y aumenta el potencial de membrana mitocondrial y la producción de ATP [9]. La hiperactivación mitocondrial por el DCA aumenta aún más la cantidad y el alcance de los radicales libres (ROS) liberados por la propia mitocondria [13]. Estos factores pueden explicar el efecto sinérgico observado al preexponer las células al DCA antes de iniciar la radiación.
Atkinson et al. [ 15] demostraron que la inhibición de la citocromo c peroxidasa y la liberación de citocromo c de las mitocondrias mitiga la muerte celular inducida por la radiación. Lee et al. [24 ] observaron una disminución de la inhibición de la muerte celular mediada por ROS mediante el bloqueo de la respuesta antioxidante dependiente del factor nuclear eritroide 2 (nrf2), lo que aumenta la radiosensibilidad de las células H1299, A549 y H640. Estos estudios demostraron la capacidad de afectar a la reacción celular a la radiación interfiriendo en procesos relacionados con el citocromo c y las ROS, lo que subraya nuestra hipótesis de que el DCA radiosensibiliza las células cancerosas a través de la activación mitocondrial que permite la liberación de ROS.
Cao et al. [21] también informaron de que el DCA actúa como radiosensibilizador en células de cáncer de próstata. Zwicker et al. [22] descubrieron que el DCA actúa como radiosensibilizador in vitro pero no in vivo en células WIDR (colorrectales) y LN18 (glioma). La falta de sinergia en el modelo in vivo de xenoinjerto WIDR se explicó por un efecto tampón del entorno, que podría haber permitido a las células tumorales mantener su programa metabólico glucolítico. Aún quedan por realizar estudios que demuestren el efecto in vivo del DCA y la radiación en el CPNM.
La RT es una terapia habitual para los pacientes con cáncer de pulmón. Sin embargo, el daño considerable que causa a los tejidos normales circundantes limita su aplicación. Durante las dos últimas décadas, los inhibidores farmacológicos de las moléculas de señalización que regulan el potencial apoptótico se han mostrado prometedores como radiosensibilizadores in vitro [25,29]. Aunque se ha descrito el uso del DCA como radiosensibilizador en el cáncer de próstata [21], hasta ahora no se ha demostrado su uso clínico en el CPNM. Aquí, mostramos la capacidad del DCA para disminuir las dosis de radiación en células A549 y H1299 NSCLC sin comprometer el nivel de efecto. Además, al demostrar que el tratamiento con DCA aumenta el consumo de oxígeno de las células A549 y H1299, hemos descubierto el potencial del DCA para actuar como activador mitocondrial. Por lo tanto, concluimos que la activación mitocondrial puede desempeñar un papel en la terapia del NSCLC cuando se integra con el tratamiento de radiación. Además, planteamos la hipótesis de que la inducción de una mayor actividad mitocondrial, es decir, una mayor liberación de radicales libres y apoptosis por el DCA, es la causa principal del potencial del DCA en la radiosensibilización de las células de NSCLC.
En resumen, demostramos que la inducción mitocondrial in vitro por el DCA radiosensibiliza significativamente las células A549 NSCLC de forma sinérgica. Esta observación justifica una evaluación adicional en un entorno in vivo.
Agradecimientos
Los autores agradecen a la Dra. Shoshana Paglin (Centro Médico Sheba) su orientación y sus fructíferas discusiones en relación con este trabajo.
Este trabajo se realizó en cumplimiento parcial de los requisitos de la tesis de doctorado de la Facultad de Medicina Sackler de la Universidad de Tel Aviv y contó con el apoyo del Centro Médico Sheba. (Sr. Ronen Shavit)
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.
REFERENCIAS
1 R. Siegel, C. DeSantis, K. Virgo, K. Stein, A. Mariotto, T. Smith, D. Cooper, T. Gansler, C. Lerro, S. Fedewa, C. Lin, C. Leach, R.S. Cannady, H. Cho, S. Scoppa, M. Hachey, R. Kirch, A. Jemal, E. Ward, Cancer treatment and survivorship statistics, 2012. CA Cancer J. Clin. 62(4), 220-241 (2012)2 Q. Wu, Y.F. Chen, J. Fu, Q.H. You, S.M. Wang, X. Huang, X.J. Feng, S.H. Zhang, Short hairpin RNA-mediated down-regulation of CENP-A attenuates the aggressive phenotype of lung adenocarcinoma cells. Cell. Oncol. 37(6), 399-407 (2014)
3 A. Koren, H. Motaln, T. Cufer, Células madre del cáncer de pulmón: una perspectiva biológica y clínica. Cell. Oncol. 36(4), 265-275 (2013)
4 N. Peled, M.W. Wynes, N. Ikeda, T. Ohira, K. Yoshida, J. Qian, M. Ilouze, R. Brenner, Y. Kato, C. Mascaux, F.R. Hirsch, Insulin-like growth factor-1 receptor (IGF-1R) as a biomarker for resistance to the tyrosine kinase inhibitor gefitinib in non-small cell lung cancer. Cell. Oncol. 36(4), 277-288 (2013)
5 M.V. Graham, J.A. Purdy, B. Emami, W. Harms, W. Bosch, M.A. Lockett, C.A. Perez, Clinical dose-volume histogram analysis for pneumonitis after 3D treatment for non-small cell lung cancer (NSCLC). Int. J. Radiat. Oncol. Biol. Phys. 45(2), 323-329 (1999)
6 H. Vakifahmetoglu, M. Olsson, B. Zhivotovsky, Death through a tragedy: mitotic catastrophe. Cell Death Differ. 15(7), 1153-1162 (2008)
7 J. Thoms, R.G. Bristow, DNA repair targeting and radiotherapy: a focus on the therapeutic ratio. Semin. Radiat. Oncol. 20(4), 217-222 (2010)
8 C. Coleman, Beneficial liaisons: radiobiology meets cellular and molecular biology. Radiother Oncol: J. Eur. Soc. Ther. Radiol. Oncol. 28(1), 1-15 (1993)
1 T. Yamamori, H. Yasui, M. Yamazumi, Y. Wada, Y. Nakamura, H. Nakamura, O. Inanami, Ionizing radiation induces mitochondrial reactive oxygen species production accompanied by upregulation of mitochondrial electron transport chain function and mitochondrial content under control of the cell cycle checkpoint. Free Radic. Biol. Med. 53(2), 260-270 (2012)
10 I. Szumiel, Ionising radiation-induced oxidative stress, epigenetic changes and genomic instability: the pivotal role of mitochondria. Int J Radiat Biol. 1-55 (2014)
11 B.A. Rupnow, S.J. Knox, The role of radiation-induced apoptosis as a determinant of tumor responses to radiation therapy. Apoptosis 4(2), 115-143 (1999)
12 R.A. Cairns, I.S. Harris, T.W. Mak, Regulation of cancer cell metabolism. Nat. Rev. Cancer 11(2), 85-95 (2011)
13 E.D. Michelakis, L. Webster, J.R. Mackey, Dichloroacetate (DCA) as a potential metabolic targeting therapy for cancer. Br. J. Cancer 99(7), 989-994 (2008)
14 X. Wang, S. Peralta, C.T. Moraes, Mitochondrial alterations during carcinogenesis: a review of metabolic transformation and targets for anticancer treatments. Adv. Cancer Res. 119, 127-160 (2013)
15 J. Atkinson, A.A. Kapralov, N. Yanamala, Y.Y. Tyurina, A.A. Amoscato, L. Pearce, J. Peterson, Z. Huang, J. Jiang, A.K. Samhan-Arias, A. Maeda, W. Feng, K. Wasserloos, N.A. Belikova, V.A. Tyurin, H. Wang, J. Fletcher, Y. Wang, I.I. Vlasova, J. Klein-Seetharaman, D.A. Stoyanovsky, H. Bayir, B.R. Pitt, M.W. Epperly, J.S. Greenberger, V.E. Kagan, A mitochondria-targeted inhibitor of cytochrome c peroxidase mitigates radiation-induced death. Nat. Commun. 2, 497 (2011)
16 S.H. Kim, Y.H. Yoo, J.H. Lee, J.W. Park, Mitochondrial NADP(+)-dependent isocitrate dehydrogenase knockdown inhibits tumorigenicity of melanoma cells. Biochem. Biophys. Res. Commun. 451(2), 246-251 (2014)
17 Z. Tatarkova, S. Kuka, M. Petras, P. Racay, J. Lehotsky, D. Dobrota, P. Kaplan, Why mitochondria are excellent targets for cancer therapy. Klin. Onkol. 25(6), 421-426 (2013)
18 S. Bonnet, S.L. Archer, J. Allalunis-Turner, A. Haromy, C. Beaulieu, R. Thompson, C.T. Lee, G.D. Lopaschuk, L. Puttagunta, G. Harry, K. Hashimoto, C.J. Porter, M.A. Andrade, B. Thebaud, E.D. Michelakis, A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11(1), 37-51 (2007)
19 N.A. Franken, H.M. Rodermond, J. Stap, J. Haveman, C. van Bree, Clonogenic assay of cells in vitro. Nat. Protoc. 1(5), 2315-2319 (2006)
20 B.K. Slinker, The statistics of synergism. J. Mol. Cell. Cardiol. 30(4), 723-731 (1998)
21 W. Cao, S. Yacoub, K.T. Shiverick, K. Namiki, Y. Sakai, S. Porvasnik, C. Urbanek, C.J. Rosser, Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Próstata 68(11), 1223-1231 (2008)
22 F. Zwicker, A. Kirsner, P. Peschke, F. Roeder, J. Debus, P.E. Huber, K.J. Weber, Dichloroacetate induces tumor-specific radiosensitivity in vitro but attenuates radiation-induced tumor growth delay in vivo. Strahlenther. Onkol. 189(8), 684-692 (2013)
23I. Papandreou, R.A. Cairns, L. Fontana, A.L. Lim, N.C. Denko, HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 3(3), 187-197 (2006)
24 S. Lee, M.J. Lim, M.H. Kim, C.H. Yu, Y.S. Yun, J. Ahn, J.Y. Song, An effective strategy for increasing the radiosensitivity of Human lung Cancer cells by blocking Nrf2-dependent antioxidant responses. Free Radic. Biol. Med. 53(4), 807-816 (2012)
25 S.J. Chmura, H.J. Mauceri, S. Advani, R. Heimann, M.A. Beckett, E. Nodzenski, J. Quintans, D.W. Kufe, R.R. Weichselbaum, Decreasing the apoptotic threshold of tumor cells through protein kinase C inhibition and sphingomyelinase activation increases tumor killing by ionizing radiation. Cancer Res. 57(19), 4340-4347 (1997)
26 V. Bhardwaj, Y. Zhan, M.A. Cortez, K.K. Ang, D. Molkentine, A. Munshi, U. Raju, R. Komaki, J.V. Heymach, J. Welsh, C-Met inhibitor MK-8003 radiosensitizes c-Met-expressing non-small-cell lung cancer cells with radiation-induced c-Met-expression. J. Thorac. Oncol. 7(8), 1211-1217 (2012)
27 E.J. Bernhard, G. Kao, A.D. Cox, S.M. Sebti, A.D. Hamilton, R.J. Muschel, W.G. McKenna, The farnesyltransferase inhibitor FTI-277 radiosensitizes H-ras-transformed rat embryo fibroblasts. Cancer Res. 56(8), 1727-1730 (1996)
28 H.J. Boeckman, K.S. Trego, J.J. Turchi, Cisplatin sensitizes cancer cells to ionizing radiation via inhibition of nonhomologous end joining. Mol. Cancer Res. 3(5), 277-285 (2005)
29 N. Balaban, J. Moni, M. Shannon, L. Dang, E. Murphy, T. Goldkorn, The effect of ionizing radiation on signal transduction: antibodies to EGF receptor sensitize A431 cells to radiation. Biochim. Biophys. Acta 1314(1-2), 147-156 (1996)
Contenido relacionado: