Sven de Mey 1, Inès Dufait 1, Heng Jiang 1, Cyril Corbet 2, Hui Wang 1, Melissa Van De Gucht 1, Lisa Kerkhove 1, Ka Lun Law 1, Hugo Vandenplas 3, Thierry Gevaert 1, Olivier Feron 2 y Mark De Ridder 1,*
1 Departamento de Radioterapia, Universitair Ziekenhuis Brussel, Vrije Universiteit Brussel, 1090 Bruselas, Bélgica; [email protected] (S.d.M.); [email protected] (I.D.); [email protected] (H.J.); [email protected] (H.W.); [email protected] (M.V.D.G.); [email protected] (L.K.); [email protected] (K.L.L.); [email protected] (T.G.)
2 Polo de Farmacología y Terapéutica (FATH), Institut de Recherche Expérimentale et Clinique (IREC), UCLouvain, 1200 Bruselas, Bélgica; [email protected] (C.C.); [email protected] (O.F.)
3 Departamento de Oncología Médica, Universitair Ziekenhuis Brussel, Vrije Universiteit Brussel, 1090 Bruselas, Bélgica; [email protected]
Correspondencia: [email protected]
Recibido: 14 de septiembre de 2020
Aceptada: 4 de diciembre de 2020
Publicado: 9 de diciembre de 2020
Resumen
El metabolismo mitocondrial es una diana atractiva para la terapia del cáncer. La reprogramación de las vías metabólicas puede potencialmente sensibilizar a los tumores con opciones de tratamiento limitadas, como el cáncer de mama triple negativo (CMTN), a la quimio y/o radioterapia. El dicloroacetato (DCA) es un inhibidor específico de la piruvato deshidrogenasa cinasa (PDK), lo que conduce a una mayor producción de especies reactivas del oxígeno (ROS). Las ROS son las principales moléculas efectoras de la radiación y un aumento de las mismas potenciará la radiorrespuesta. En este estudio, evaluamos los efectos del DCA y la radioterapia en dos líneas celulares de TNBC, EMT6 y 4T1, en condiciones aeróbicas e hipóxicas. Como era de esperar, el tratamiento con DCA redujo la piruvato deshidrogenasa fosforilada (PDH) y disminuyó tanto la tasa de acidificación extracelular (ECAR) como la producción de lactato. Sorprendentemente, el tratamiento con DCA provocó un aumento significativo de la producción de ROS (hasta 15 veces) en las células cancerosas hipóxicas, pero no en las aeróbicas. De forma consistente, el DCA radiosensibilizó las células tumorales hipóxicas y los esferoides 3D, dejando inalterada la radiosensibilidad intrínseca de las células tumorales. Nuestros resultados sugieren que, aunque descrito como un fármaco promotor de la fosforilación oxidativa (OXPHOS), el DCA también puede aumentar las radiorrespuestas hipóxicas. Por lo tanto, este estudio allana el camino para dirigir el metabolismo mitocondrial de las células cancerosas hipóxicas, en particular para combatir la radiorresistencia.
Palabras clave: dicloroacetato; radiosensibilidad hipóxica; cáncer de mama; especies reactivas del oxígeno
2020 por los autores. Licenciatario MDPI, Basilea, Suiza. Este artículo es un artículo de acceso abierto distribuido bajo los términos y condiciones de la licencia Creative Commons Attribution (CC BY) (http://creativecommons.org/licenses/by/4.0/).
INTRODUCCIÓN
El cáncer de mama es el cáncer más frecuente en las mujeres de todo el mundo y causa anualmente 627.000 muertes [1]. En las últimas décadas se han realizado avances significativos en el tratamiento del cáncer de mama. Sin embargo, sólo se dispone de tratamientos limitados para las pacientes con cáncer de mama triple negativo/basal-like [2,3,4]. El tratamiento estándar de los cánceres de mama de alto riesgo consiste en quimioterapia neoadyuvante y cirugía, seguidas de irradiación postoperatoria de todo el pecho/pared torácica. En la actualidad, los investigadores se centran en el hipofraccionamiento de la radioterapia adyuvante (ensayo FAST-Forward [5] ) o en la combinación de quimioterapia con radioterapia preoperatoria. La radioterapia preoperatoria podría mejorar la supervivencia libre de enfermedad y la calidad de vida [6,7,8,9,10,11].
El principal efecto de la radiación, en particular de la radiación de baja transferencia de energía lineal, es la inducción de especies reactivas del oxígeno (ROS). Durante la radioterapia, se crean ROS por la radiólisis del agua en entornos extracelulares, que son tóxicas para las células tumorales y el tejido normal cercano. Alrededor de dos tercios de los daños en el ADN inducidos por la radiación se atribuyen a las ROS en células de mamíferos [12]. La respuesta de las células al daño del ADN inducido por la radiación depende en gran medida de la presencia de oxígeno. En efecto, las moléculas de oxígeno pueden fijar los daños en el ADN producidos por los radicales libres. Esto se conoce como la «hipótesis de la fijación del oxígeno» [12,13]. En ausencia de oxígeno, los radicales del ADN son reducidos por compuestos que contienen grupos sulfhidrilo, que reparan el ADN a su forma original. Siguiendo esta hipótesis, la hipoxia, definida por bajos niveles de oxígeno en el tumor, es una de las principales causas de fracaso clínico de la radioterapia [14,15]. La hipoxia es una característica común del microambiente tumoral. Las ERO y la hipoxia son dos factores con efectos opuestos en la radiorrespuesta del tumor [16]. La hipótesis generalmente aceptada afirmaba que en las regiones hipóxicas del tumor se producía un menor estrés oxidativo debido a la escasez de oxígeno sustrato de las ROS. Sin embargo, pruebas recientes revelan que, en condiciones de hipoxia, las células generan más ROS, principalmente a través del metabolismo mitocondrial [17,18,19,20].
Un rasgo distintivo de las células tumorales es la capacidad de alterar su metabolismo, proporcionándoles la energía y los metabolitos necesarios para su crecimiento y supervivencia en condiciones limitadas de nutrientes y oxígeno. Sin embargo, en presencia de O2, las células cancerosas también adaptan su metabolismo hacia la glucólisis, desviando la oxidación mitocondrial del piruvato hacia la producción de lactato [21,22]. Este efecto se conoce como efecto Warburg. Informes recientes indican que el efecto Warburg está implicado en la resistencia al estrés citotóxico inducido por la quimioterapia o la radioterapia [23,24,25,26,27]. De este modo, los métodos de tratamiento que bloquean o reducen el metabolismo glucolítico pueden aumentar la sensibilidad de las células tumorales a la radioterapia.
En condiciones de hipoxia, el factor inducible por hipoxia 1-alfa (HIF1α) provoca un aumento de la expresión de las piruvato deshidrogenasa quinasas (PDK1-4) [28]. Estas enzimas son responsables del cambio de metabolismo en las mitocondrias mediante la regulación del estado de fosforilación (es decir, el estado de actividad) de la piruvato deshidrogenasa (PDH), que es una importante proteína de control entre la glucólisis y la fosforilación oxidativa mitocondrial (OXPHOS). El dicloroacetato (DCA), una molécula pequeña inhibidora de la PDK, puede invertir el efecto Warburg activando la PDH y reorientando el metabolismo del piruvato hacia la mitocondria. La inhibición de la PDK mediante DCA se utiliza para tratar la acidosis láctica y las enfermedades mitocondriales hereditarias [29,30]. En conjunto, estas observaciones han llevado a considerar el DCA como un fármaco potencial contra el cáncer [30,31].
Se ha demostrado que el DCA aumenta la radiosensibilidad de las células de cáncer de colon y próstata, así como de los tumores de carcinoma esofágico de células escamosas y glioblastoma [32 ,33,34,35]. Sin embargo, no se han realizado investigaciones similares en células de cáncer de mama. El principal mecanismo de radiosensibilización en estos modelos se atribuyó al estrés oxidativo. Al mismo tiempo, la detención del ciclo celular en la fase G2-M y la reducción de la capacidad de reserva mitocondrial también contribuyeron a los efectos radiosensibilizadores. Basándose en los resultados descritos anteriormente, actualmente se están llevando a cabo dos ensayos clínicos (uno en carcinoma de cabeza y cuello y otro en glioblastoma) en los que se investigan los efectos antitumorales del tratamiento combinado de DCA con radioterapia [36,37]. Todas las investigaciones preclínicas se han realizado en condiciones aeróbicas, pero no se dispone de datos sobre los efectos del tratamiento con DCA en condiciones hipóxicas. Por lo tanto, en el presente estudio, primero examinamos la hipótesis de que el DCA reduce el lactato y cambia el metabolismo de un fenotipo glucolítico a OXPHOS. A continuación, determinamos si el DCA puede radiosensibilizar las células hipóxicas del cáncer de mama y examinamos los mecanismos subyacentes. Los resultados de este estudio pueden tener importantes implicaciones para los ensayos clínicos destinados a utilizar inhibidores de la PDK para mejorar la radiorrespuesta en pacientes con cáncer de mama.
Resultados
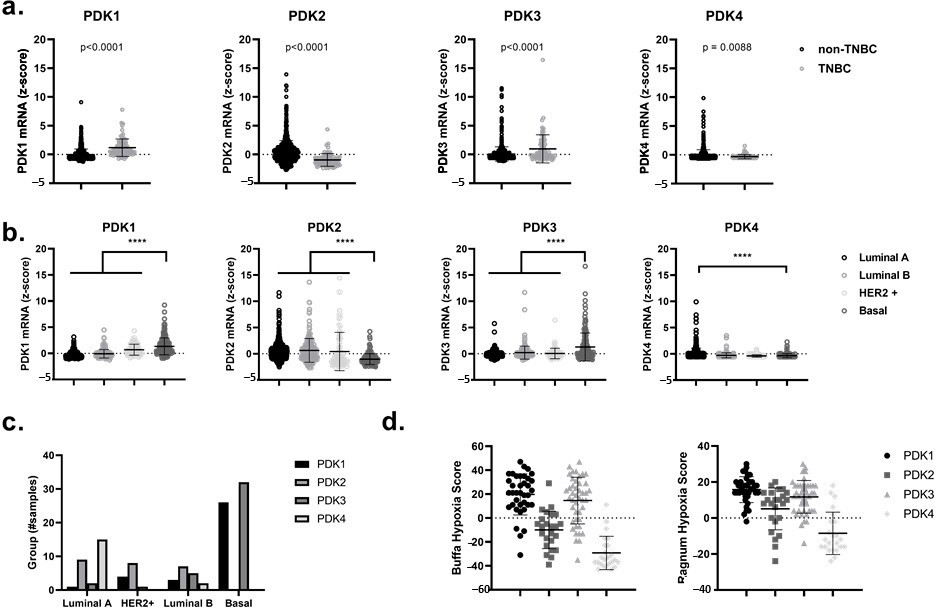
Laalta expresión de PDK1 y PDK3 en pacientes con cáncer de mama triple negativo (CMTN) y cáncer de mama de tipo basal está correlacionada con una firma génica relacionada con la hipoxia
En primer lugar, utilizando el cBioPortal for Cancer Genomics en línea y de acceso público, analizamos los niveles de ARNm de los cuatro isómeros diferentes de PDK, a saber, PDK1, PDK2, PDK3 y PDK4, en datos derivados de pacientes del conjunto de datos de tumores primarios de cáncer de mama TCGA (PanCancer Atlas and Cell 2015) [38,39]. Mostramos un aumento significativo (p < 0,0001) en la expresión de PDK1 y PDK3 en TNBC frente a no TNBC y en cánceres de mama de tipo basal en relación con otros subtipos como los cánceres de mama luminales A, luminales B y enriquecidos con HER2 (Figura 1a-c). En particular, la regulación al alza de PDK1 y PDK3 en pacientes con cáncer de mama podría correlacionarse con puntuaciones de hipoxia más altas de Ragnum y Buffa [40,41] (Figura 1d). Estas puntuaciones de hipoxia se basan en la expresión diferencial de genes específicos relacionados con la hipoxia y son de libre acceso a través de cBioPortal for Cancer Genomics. La regulación al alza observada de PDK1 y PDK3 en cánceres de mama de tipo basal y su correlación con un fenotipo hipóxico (que está vinculado a la activación de un programa transcripcional dependiente de HIF1α), sugiere que el uso de inhibidores de PDK podría constituir una modalidad terapéutica atractiva con fines de radiosensibilización hipóxica [42,43,44].
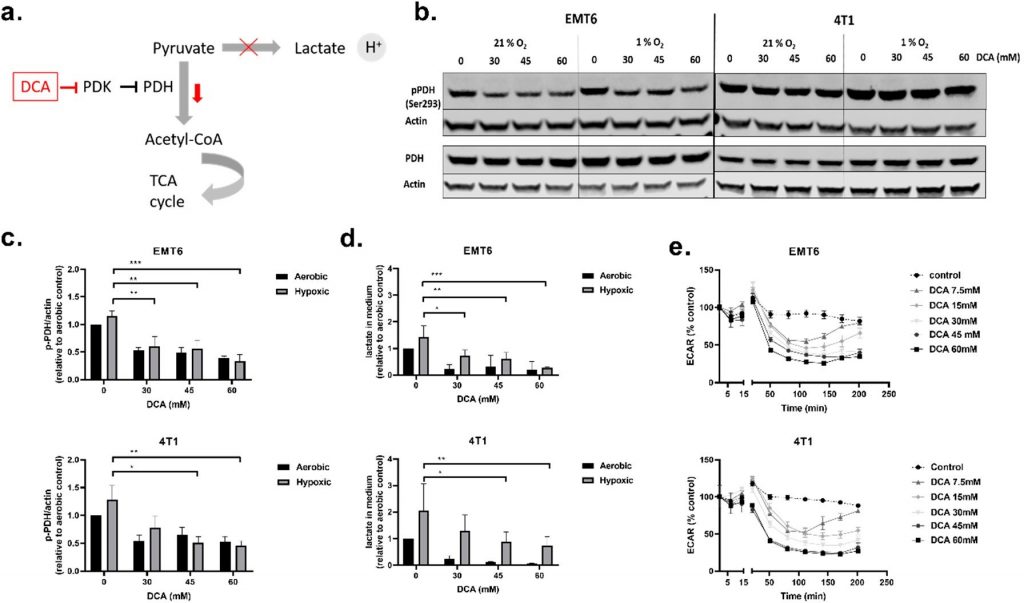
El DCAdisminuyó la PDH fosforilada, los niveles de lactato extracelular y la tasa de acidificación extracelular (ECAR)
Comenzamos nuestros experimentos in vitro realizando ensayos de viabilidad (Figura S1a,b) para determinar las propiedades inhibidoras del crecimiento del DCA. El DCA redujo la viabilidad celular de forma dependiente de la dosis en las células cancerosas EMT6 y 4T1, independientemente del estado del O2 (Figura S1a). Un ensayo de proliferación también mostró que el aumento de las concentraciones de DCA nos llevó a observar un cambio de retraso del crecimiento a efectos citostáticos e incluso citotóxicos (Figura S1b).
A continuación, investigamos la influencia del DCA sobre la actividad de PDK1-4 y el metabolismo de las células TNBC. La PDH es una proteína clave de la glucólisis y la OXPHOS mitocondrial, lo que significa que la PDH cataliza la descarboxilación limitante de piruvato en acetil-CoA. La PDH es inhibida a través de la fosforilación (en Ser293) por la PDK, y esta inhibición puede ser revertida a través de la desfosforilación por la piruvato deshidrogenasa fosfatasa (PDP) [45,46] (Figura 2a). A dosis de toxicidad aceptables (30 mM, 45 mM y 60 mM), evaluamos el efecto del DCA sobre la actividad PDK midiendo los niveles de PDH fosforilada (p-PDH), lactato en el medio extracelular y el ECAR de las células en tiempo real (Figura 2b-e). Las tres dosis de DCA disminuyeron la cantidad de p-PDH en las células EMT6 y 4T1 de forma dosis-dependiente tanto en condiciones oxigenadas como hipóxicas. La disminución de la p-PDH fue significativa para todas las dosis de DCA en EMT6, mientras que en 4T1, sólo 45 mM y 60 mM disminuyeron significativamente la p-PDH en condiciones de hipoxia (Figura 2b,c). Al evaluar el efecto de dosis menores de DCA, observamos que en las células EMT6 la cantidad de p-PDH empieza a disminuir cuando se tratan con 3 mM de DCA y en las células 4T1 cuando se tratan con 10 mM de DCA (Figura S2a,b). La cantidad de lactato en el medio se elevó en condiciones hipóxicas en comparación con las condiciones aeróbicas en ambas líneas celulares, lo que apoya un aumento neto del recambio glucolítico en células privadas de O2 [47,48]. En concordancia con los resultados de Western blot, el tratamiento con DCA condujo a una disminución dependiente de la dosis de lactato en el medio para ambas líneas celulares (Figura 2d). Aunque la reducción de la liberación de lactato fue drástica en condiciones aeróbicas, también se observó una reducción dependiente de la dosis en la producción del producto glicolítico final en condiciones de hipoxia. Por último, el DCA a partir de una dosis de 7,5 mM, provocó una reducción dependiente del tiempo del ECAR tanto en EMT6 como en 4T1 (Figura 2e). La caída inicial del ECAR tras el tratamiento con dosis inferiores a 30 mM de DCA se compensó al cabo de 2,5 h en EMT6. En 4T1, observamos el mismo efecto con dosis de DCA inferiores a 15 mM. Estos resultados indican que el tratamiento de líneas celulares TNBC murinas con DCA inhibe la fosforilación de PDH y reduce el alcance de la glucólisis tanto en condiciones aeróbicas como hipóxicas.
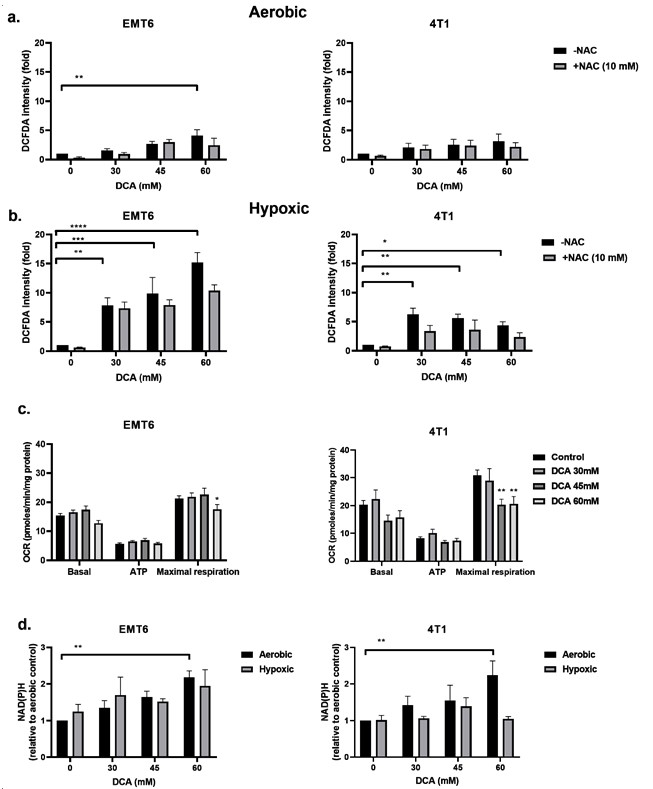
ElDCA induce la producción de ROS en células cancerosas hipóxicas
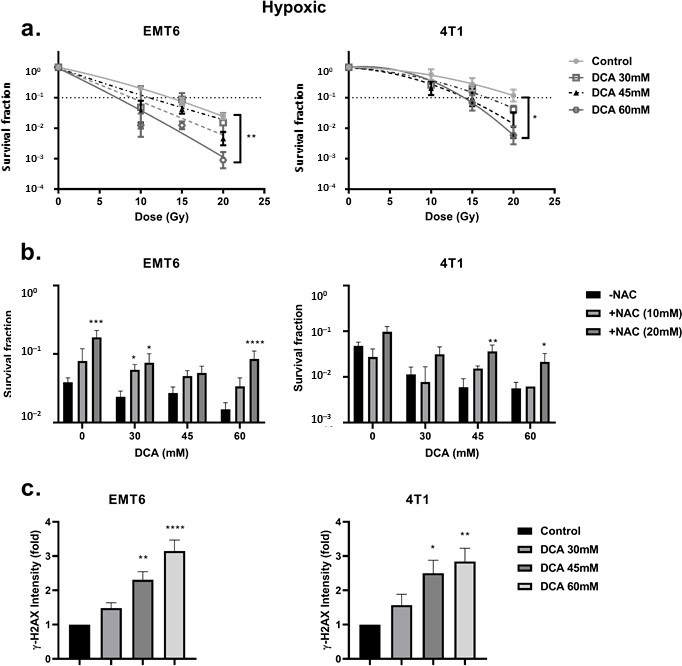
Se ha descrito que la inhibición de la PDK y la activación de la PDH están asociadas a la regulación al alza de las ROS intracelulares [49,50]. Las ROS tienen una importancia fundamental en la generación de daños en el ADN tras la radiación. Examinamos los niveles de ROS en las células EMT6 y 4T1 en condiciones aeróbicas e hipóxicas utilizando la sonda CM-H2DCFDA. Como se muestra en la Figura 3a,b, el DCA desencadenó una producción de ROS dependiente de la dosis tanto en EMT6 como en 4T1. En condiciones aeróbicas, sólo la dosis más alta (60 mM) de DCA generó un aumento significativo de ROS de hasta cinco veces en comparación con el control para las células EMT6. En las células 4T1, no se detectó un aumento significativo de ROS en condiciones aeróbicas. El aumento de ROS se contrarrestó en parte mediante la adición del eliminador de ROS N-acetilcisteína (NAC). En condiciones de hipoxia, se observó un aumento de las ERO dependiente de la dosis, hasta 15 veces en las células EMT6 y hasta 5 veces en las células 4T1; la dosis más baja de DCA produjo un aumento significativo de las ERO en ambos tipos celulares (Figura 3b).
Pensamos que el aumento de la producción de ROS en condiciones de hipoxia podría deberse a los efectos combinados de la alteración de la cadena de transporte de electrones mitocondrial (debido a la reducción del O2 como aceptor final de electrones) y el metabolismo oxidativo del piruvato forzado por el DCA. Utilizando el analizador Seahorse, descubrimos que el DCA no tenía ningún impacto sobre la respiración basal y la producción de ATP, pero disminuía significativamente la capacidad respiratoria máxima en las células tumorales EMT6 y 4T1 (Figura 3c). Otra posibilidad es que el DCA aumente los niveles de NAD(P)H, lo que está relacionado con una mayor producción de ROS. Observamos que, en condiciones aeróbicas, el tratamiento con DCA producía un aumento dependiente de la dosis de NAD(P)H en las células EMT6 y 4T1 (Figura 3d). En condiciones de hipoxia, demostramos un posible aumento de NAD(P)H en las células EMT6 tratadas con 60 mM de DCA, pero ningún aumento en las células 4T1 (Figura 3d). Junto con los datos anteriores sobre ROS, estos hallazgos sugieren que, aunque el metabolismo mitocondrial sigue preservado, un aumento local de la producción de ROS tras la exposición al DCA puede alterar la integridad y la función de las mitocondrias.
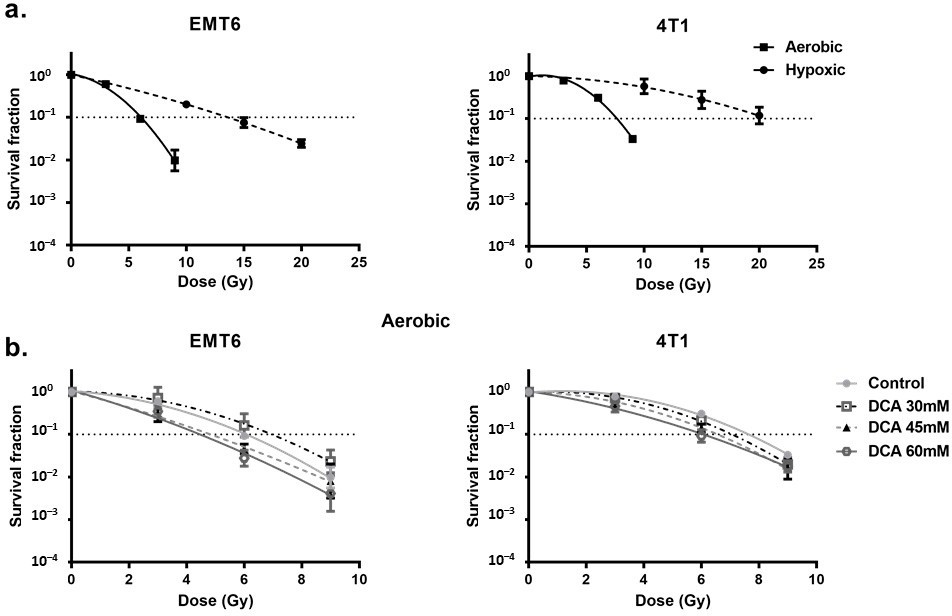
ElDCA radiosensibiliza las células hipóxicas mediado por ROS
En primer lugar, observamos la radioresistencia de las células inducida por la hipoxia. Encontramos una radiorrespuesta gravemente alterada al comparar las condiciones hipóxicas con las aeróbicas, con una relación de aumento de oxígeno de 2,7 y 2,3 para las células tumorales EMT6 y 4T1, respectivamente (Figura 4a). En este escenario, observamos que el tratamiento con DCA causaba un pequeño efecto de radiosensibilización intrínseca en las células EMT6 pero no en las 4T1 (Figura 4b). Curiosamente, en consonancia con los resultados de la generación de ROS en condiciones de hipoxia, el DCA 60 mM superó significativamente (p < 0,05) la radiorresistencia hipóxica con ratios de mejora de 2,3 y 1,5 a 60 mM para las células tumorales EMT6 y 4T1, respectivamente (Figura 5a). El efecto radiosensibilizador fue revertido por la NAC tanto en las células EMT6 como en las 4T1 (Figura 5b). La causa principal de la muerte celular inducida por radiación por ROS es a través de la inducción de roturas de doble cadena en el ADN (ds-ADN) [12,51]. Por lo tanto, examinamos el daño ds-DNA tras el tratamiento con DCA cuantificando el estado de fosforilación de γH2AX en condiciones de hipoxia. Como se muestra en la Figura 5c, el DCA incrementó la formación de daño ds-DNA tanto en EMT6 como en 4T1 de forma dosis-dependiente.
ElDCA radiosensibiliza los cultivos celulares tridimensionales (esferoides)
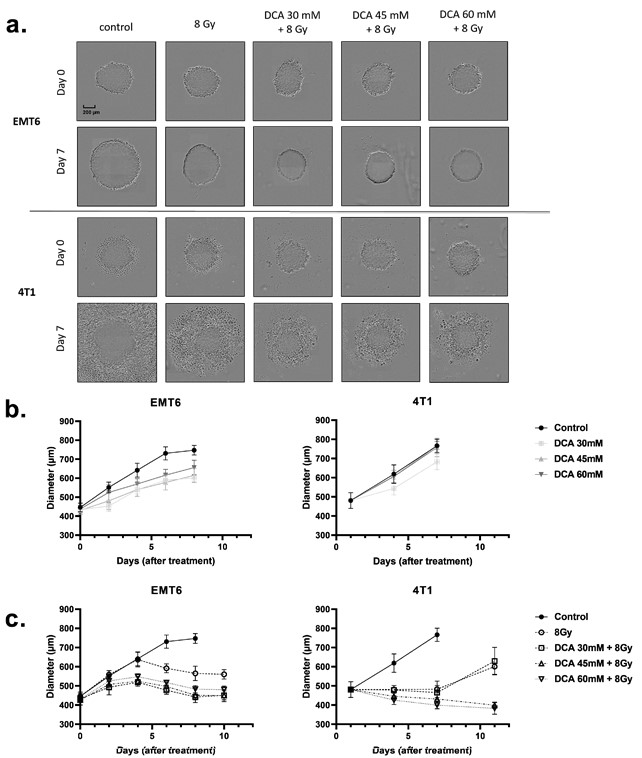
Los hallazgos anteriores nos llevaron a examinar si el DCA también podría mejorar la radiosensibilidad de los modelos de cultivos celulares tridimensionales (3D) (Figura 6a-c) que imitan mejor las propiedades fisicoquímicas del microambiente tumoral, incluidos los gradientes de oxígeno. Utilizando esferoides obtenidos a partir de cultivos celulares EMT6 y 4T1, medimos el crecimiento de los esferoides tras el tratamiento con DCA y radioterapia (Figura 6a). El tratamiento con DCA provocó por sí solo un pequeño retraso en el crecimiento de las células EMT6, pero no alteró el crecimiento de los esferoides 4T1 (Figura 6b). Una irradiación de 8 Gy redujo el crecimiento de los esferoides tumorales, efecto acentuado por la combinación con el tratamiento con DCA (Figura 6a,c). Cabe destacar que, mientras que en los esferoides 4T1 se observaron efectos citotóxicos (revelados por halos de células muertas), en los esferoides EMT6 se observaron efectos citostáticos (Figura 6a).
La combinación de DCA y radioterapia no retrasa el crecimiento tumoral in vivo
A continuación examinamos si el beneficio in vitro de la combinación de DCA y radioterapia podía validarse in vivo (Figura S3a-d). Ratones inyectados con células de cáncer de mama EMT6 o 4T1 fueron expuestos a radiación de fracción única (12 Gy o 15 Gy, respectivamente) (Figura S3a,c) o fraccionada (5*4 Gy o 5*6 Gy, respectivamente) (Figura S3b,d). Las dosis de radiación única y fraccionada por tipo tumoral son similares en dosis biológica efectiva (BED) y difieren en función de la radiosensibilidad intrínseca de las líneas celulares utilizadas. Cabe destacar que la inyección de DCA i.p. o i.t. durante 10 días resultó segura sin inducir toxicidad apreciable (Figura S4a-d). La radiación sola retrasó el crecimiento tumoral en EMT6 durante siete días con una sola fracción y cuatro días para la radiación fraccionada (Figura S3a,b). En los tumores 4T1, la radiación retrasó el crecimiento tumoral con cinco días para la fracción única y 10 días para la radiación fraccionada, como era de esperar (Figura S3c,d). El DCA (300 mg/kg), mediante inyección intraperitoneal (ip) o intratumoral (it), no retrasó el crecimiento tumoral y tampoco lo hizo la combinación de DCA con radiación (Figura S3a-d). A continuación, validamos si el tratamiento con DCA podía inducir hipoxia en los tumores (Figura S3e,f). Aunque la extensión de la hipoxia teñida con pimonidazol no se vio alterada en los tumores EMT6, se observó una tendencia a la disminución de la hipoxia en respuesta al DCA en los tumores 4T1.
Discusión
El objetivo de este estudio era examinar si el DCA dirigido al metabolismo mitocondrial podría sensibilizar las células de cáncer de mama TNBC/basal-like a la radioterapia. La mayoría de los TNBC y cánceres de mama basales son tumores agresivos para los que las opciones de tratamiento son limitadas y el pronóstico es malo [2,3,4]. Estos tumores presentan un fenotipo glucolítico aumentado que favorece su mal pronóstico y se correlaciona además con la radioresistencia [52]. En el presente estudio, encontramos que los niveles de ARNm de dos de las cuatro isoformas de PDK (PDK1 y PDK3) están regulados al alza en los subtipos de cáncer de mama TNBC y basal-like. La sobreexpresión de las PDK se ha detectado en múltiples muestras de tumores humanos [42,53,54,55,56,57,58,59,60,61], y muchas líneas celulares de cáncer presentan un aumento sustancial de las isoformas PDK [50,62,63]. Se ha descrito que la sobreexpresión de PDK está asociada a un mal pronóstico en diversos tipos de tumores [53,54,55,56,57,58,59,60,61]. La sobreexpresión de PDKs en células cancerosas está influenciada por varios factores de transcripción, como HIF1 [28 ,64]. HIF1 suprime activamente la OXPHOS transactivando directamente los genes que codifican PDK1 y PDK3. A su vez, las PDK fosforilan e inactivan la PDH [42]. Así pues, la regulación al alza de las PDK en el cáncer puede atribuirse instantáneamente tanto a las mutaciones transformadoras como al microambiente hipóxico del tumor. En consonancia con estos hallazgos, observamos que la regulación al alza del ARNm de PDK1 y PDK3 está correlacionada con perfiles génicos relacionados con la hipoxia. Por lo tanto, la reprogramación metabólica de las enzimas PDK para pasar de la glucólisis a la OXPHOS parece una vía terapéutica prometedora para tratar los cánceres de mama con opciones terapéuticas limitadas.
El principal hallazgo de nuestro estudio es que el inhibidor de PDK DCA puede reducir la actividad glucolítica de las células de cáncer de mama en presencia de oxígeno, pero también en condiciones de hipoxia [29]. Encontramos que el DCA disminuye la cantidad de PDH fosforilada y desencadena una disminución dependiente de la dosis en los niveles de lactato extracelular y ECAR en células aeróbicas e hipóxicas. A continuación, combinamos DCA y radioterapia bajo la hipótesis de que al invertir el fenotipo glucolítico y dirigir más piruvato hacia la oxidación mitocondrial, las células tumorales podrían producir más ROS y volverse más sensibles a la radiación. De hecho, se han descrito efectos radiosensibilizadores intrínsecos del DCA en células de glioblastoma [34,35], células de carcinoma pulmonar de células no pequeñas (CPCNP) [65,66], células de cáncer colorrectal [35], células de cáncer de próstata [32] y células de meduloblastoma resistentes a la radiación [67]. Los mecanismos propuestos son la detención del ciclo celular en la fase G2-M, la creación de daños adicionales en el ADN y la muerte celular consecutiva en respuesta al aumento de la producción mitocondrial de ROS. En el presente estudio, encontramos que mientras que se observaron efectos radiosensibilizantes mínimos con la concentración no tóxica más alta de DCA en condiciones aeróbicas, el DCA radiosensibilizó fuertemente las células de cáncer de mama hipóxicas, tanto en esferoides 2D como 3D. Aunque en teoría las ERO están asociadas a mecanismos oxidativos, existe una asociación entre la hipoxia y las ERO en los tumores. La hipoxia aumenta la generación de ROS mediante la prolongación de la vida útil de los radicales semiquinona; recíprocamente, las ROS ayudan a las células tumorales a adaptarse a la hipoxia mediante la estabilización de HIF1-α [16,68]. Sin embargo, las agresiones extracelulares pueden romper esta asociación desencadenando una producción excesiva de ROS, lo que afecta a la respiración mitocondrial y, por tanto, disminuye la fracción hipóxica en los tumores [16,69,70]. En este contexto, el trióxido de arsénico inhibe el consumo de oxígeno de las células tumorales a través de un aumento de las ROS intracelulares, lo que conduce a una mayor radiorrespuesta [71]. También se ha descrito que la supresión de la glucólisis aumenta la radiorrespuesta. Esto puede hacerse mediante ritonavir (inhibidor del transportador de glucosa), 2-deoxiglucosa (inhibidor de la hexoquinasa) y lonidamina (inhibidor de la hexoquinasa), que se están investigando en ensayos clínicos en diferentes tipos de cáncer [72,73,74,75]. Otra posibilidad es que se creen ROS tras el tratamiento con DCA debido a la inducción de la NADPH oxidasa [76]. Sin embargo, no se encontraron pruebas directas para concluir que el DCA puede aumentar la NADPH oxidasa [77]. Observamos un aumento dependiente de la dosis de NAD(P)H en condiciones aeróbicas, pero no en condiciones hipóxicas en las células EMT6 y 4T1. Por lo tanto, nuestra hipótesis es que el aumento de NAD(P)H y la regulación al alza de las NADPH oxidasas sólo desempeñan un papel menor en el aumento de ROS en condiciones de hipoxia. En nuestras manos, es muy probable que el mecanismo primario de los efectos de radiosensibilización observados resulte de los aumentos de muchas veces en la producción de ROS (hasta 15 veces) después del tratamiento con DCA en condiciones de hipoxia.
En línea con la literatura, hemos demostrado la necesidad de concentraciones suprafisiológicas de DCA para provocar alteraciones en la actividad metabólica, aumento de la formación de ROS y radiosensibilización [29]. Nuestra hipótesis central es que estos efectos son consecuencia de la inhibición de la PDK producida por el DCA [78]. Sin embargo, las concentraciones requeridas para inducir los resultados medidos son varias veces superiores a la constante de inhibición (Ki) de PDK1-4. Cabe destacar que el DCA existe fisiológicamente como un anión, es relativamente impermeable a la membrana a pesar de su pequeño tamaño y por lo tanto requiere el transportador mitocondrial de piruvato para su captación mitocondrial [79,80]. Sin embargo, la conjugación del DCA con un transportador lipofílico mejoró el transporte mitocondrial. Esto redujo el valor IC50 del DCA del rango milimolar al rango micromolar bajo, que está dentro del rango Ki de la PDK1-4 [81]. El DCA imita la inhibición efectiva de PDK1-4 por siRNA, y el DCA añadido al siRNA de PDK no tuvo efectos adicionales [49,50,60,82,83,84,85,86,87,88]. Además, una molécula pequeña como el DCA podría afectar directa o indirectamente a otras dianas celulares y moleculares. Investigaciones recientes descubrieron que el tratamiento con DCA aumentaba la concentración de cada intermediario del TCA pero no afectaba a la captación de glucosa ni a la glucólisis [89]. Otros investigadores mostraron evidencias que sugieren que el DCA puede aumentar la biosíntesis de novo de CoA. Dado que altas concentraciones de CoA pueden ser tóxicas para las células, este efecto metabólico podría ser parcialmente responsable de la toxicidad de las células cancerosas mediada por el DCA [90]. Investigaciones recientes introdujeron una hipótesis novedosa, sugiriendo que la eficacia del DCA contra el cáncer podría derivarse de su capacidad para antagonizar el acetato. Unos niveles elevados de acetato pueden potenciar la síntesis de ADN, ARN y proteínas. Además, puede estar asociado con la resistencia a los fármacos contra el cáncer [91]. Por último, los investigadores han descubierto que el DCA puede activar la vía de señalización AMPK, lo que conduce a una cascada de efectos metabólicos y anticancerígenos [92,93]. No obstante, nuestra hipótesis sigue siendo que, en condiciones de hipoxia, la entrada de piruvato en las mitocondrias provoca un aumento de los niveles de ROS, lo que radiosensibiliza las células cancerosas. No pudimos recapitular estos efectos in vivo, y se necesita más trabajo para determinar cómo traducir los efectos radiosensibilizadores del DCA en compartimentos tumorales hipóxicos. Los problemas farmacocinéticos relacionados con la administración de DCA in vivo pueden ser excluidos, y la dosis de DCA utilizada (150 mg/kg) está bien dentro de las dosis utilizadas en la literatura [29]. La dosis equivalente en humanos de nuestra dosis de DCA utilizada in vivo es de 12 mg/kg/d, bien dentro de la zona tolerable utilizada en los ensayos clínicos. Una posible explicación del fracaso de nuestros experimentos in vivo es que se necesita una dosis mayor de DCA para tener un efecto radiosensibilizador in vivo. De hecho, en los últimos 30 años, el DCA se ha administrado con un éxito razonable como fármaco en investigación para el tratamiento de la diabetes tipo 2, la hiperlipoproteinemia adquirida y congénita, la isquemia miocárdica, la acidosis láctica adquirida y congénita y, más recientemente, el cáncer [29,30]. Varios ensayos de fase I/II están investigando la seguridad del DCA y su actividad como agente anticancerígeno. El DCA se absorbe rápidamente y puede incluso atravesar la barrera hematoencefálica. Dos ensayos de fase I examinaron la seguridad del DCA oral en pacientes con tumores cerebrales malignos recurrentes o metástasis cerebrales de cánceres no relacionados con el sistema nervioso central [94,95]. Estos estudios indicaron que el DCA es generalmente bien tolerado por los pacientes. Una explicación alternativa para la desconcertante diferencia entre los efectos in vitro e in vivo es, en realidad, más probable que esté relacionada con los cambios en los fenotipos metabólicos cuando las células cancerosas se inyectan (ectópicamente) in vivo. De hecho, el aumento de la radiosensibilidad in vitro por DCA se consigue en condiciones de hipoxia y alto metabolismo glucolítico, de modo que el cambio de glucólisis a OXPHOS puede observarse en el tratamiento con DCA junto con un aumento adicional de ROS.
La limitada extensión de la hipoxia en tumores in vivo puede reflejar una capacidad restringida del DCA para inducir un cambio que ya está presente en tumores bien oxigenados que dependen en gran medida de la OXPHOS. Se justifican nuevas investigaciones para comprobar los efectos radiosensibilizadores del DCA en modelos de tumores de mama de ratón caracterizados por una angiogénesis limitada y fracciones hipóxicas elevadas.
En conclusión, demostramos que el DCA supera la radiorresistencia hipóxica de las células de cáncer de mama en sistemas 2D y 3D, lo que puede atribuirse principalmente al aumento de ROS. Sorprendentemente, el cambio inducido por el DCA en el metabolismo de glucólisis a OXPHOS también crea estrés oxidativo en condiciones de hipoxia. El DCA se ha utilizado durante muchos años para tratar afecciones metabólicas y enfermedades mitocondriales hereditarias. En la última década, el DCA se ha reutilizado en gran medida como fármaco anticancerígeno con datos preclínicos prometedores, informes de casos y ensayos clínicos, como se ha descrito anteriormente. Los resultados preclínicos actuales indican que es necesario seguir investigando el potencial anticancerígeno del DCA, teniendo en cuenta que las células tumorales hipóxicas no se libran del inhibidor de la PDK, que puede inducir un estrés oxidativo letal, en particular cuando se combina con radioterapia.
Materiales y métodos
Análisis de cohortes de cáncer de mama TCGA
Los perfiles de expresión de ARNm de PDK1-4 (RNA Seq V2 RSEM o log RNA Seq V2 RSEM) se consultaron en el sitio web cBioPortal en forma de datos transformados en z-score [38,39]. Los datos consultados se evaluaron a partir de 1084 casos de cáncer de mama disponibles públicamente del TCGA PanCancer Atlas y de 817 casos de cáncer de mama de la base de datos TCGA Cell 2015. Para el conjunto de datos TCGA Cell 2015, el análisis de PDK1-4 se realizó comparando la subpoblación triple negativa con el resto de casos de cáncer de mama. El cáncer de mama triple negativo se determinó mediante un estado «negativo» para las puntuaciones inmunohistoquímicas de los genes del receptor de estrógeno (RE), el receptor de progesterona (RP) y el receptor 2 del factor de crecimiento epidérmico humano (HER2) (un total de 83 casos). En la base de datos TCGA PanCancer Atlas, el análisis de la expresión de PDK1-4 se realizó en diferentes categorías Pam50. Pam50 es una firma de 50 genes que clasifica el cáncer de mama en cinco subtipos moleculares intrínsecos: Luminal A, Luminal B, HER2-enriched, Basal-like y Normal-like. Los datos clínicos y de expresión de ARNm del cáncer de mama del TCGA se analizaron directamente en el sitio web cBioPortal o se descargaron para su posterior análisis. El análisis de la puntuación de hipoxia de Buffa y Ragnum de las muestras en las que PDK1-4 tiene una expresión de una puntuación z superior a 2 se realizó directamente en el sitio web cBioPortal.
Líneas celulares y productos químicos
La línea celular del adenocarcinoma mamario murino EMT6 fue cedida amablemente por Edith Lord (University of Rochester, Cancer Center, Nueva York), y las células 4T1 se obtuvieron de American Type Culture Collection. Todos los experimentos se realizaron en medio Roswell Park Memorial Institute 1640 (Thermo Fisher Scientific, Waltham, MA, EE.UU.) suplementado con un 10% de suero bovino fetal (Greiner Bio-One, Kremsmünster, Austria). Se utilizó tampón HEPES para todos los tratamientos de las células. Los productos químicos se obtuvieron de Sigma-Aldrich (Sigma-Aldrich, St. Louis, MO, EE.UU.) a menos que se indique lo contrario
Tratamientos
EMT6 y 4T1 se cultivaron hasta confluencia y se trataron con DCA durante 16 h a las concentraciones indicadas. Se añadió N-acetil cisteína (NAC) a 10 mM o 20 mM a los cultivos 1 h antes y durante el tratamiento con DCA. A continuación, los cultivos se utilizaron para los análisis que se describen a continuación. El tratamiento se realizó en condiciones aeróbicas o hipóxicas. La hipoxia se indujo mediante la incubación en un gas equilibrado de nitrógeno y dióxido de carbono que contenía un 1% de oxígeno [96]
Ensayo MTT <p><strong>Western Blot<br></strong>Los análisis Western blot se realizaron como se ha descrito previamente <sup><a href=»#99″>[99]</a></sup>. Brevemente, las células se lisaron en tampón Triton-X al 1% suplementado con un inhibidor de la fosfatasa (P5726), un inhibidor de la proteasa (P8340) y trifluoroacetato de leupeptina (L2023). Se centrifugaron los lisados y se determinó la concentración de proteínas mediante el ensayo de proteínas DC de Bio-Rad (Bio-Rad 500-0116). Se cargaron cantidades equivalentes de proteínas en un gel de acrilamida de resolución del 12%. La transferencia de proteínas se realizó durante la noche a 4 °C utilizando una membrana de nitrocelulosa (0,45 µM, Thermo 88018, Thermo Fisher Scientific). Las membranas se bloquearon con BSA al 5% en TBS y se lavaron (TBST). Las membranas bloqueadas se marcaron con anticuerpos primarios durante la noche a 4 °C. Los anticuerpos primarios se marcaron con anticuerpos secundarios en el infrarrojo cercano (IRDyes 680 RD o 800 CW, LI-COR Biosciences, Lincoln, NE, EE.UU.), se detectaron y cuantificaron con el sistema Odyssey Fc Imaging System (LI-COR Biosciences). Los anticuerpos primarios fueron: fosfo-PDH (ABS204, MERCK, Darmstadt, Alemania), PDH total (C54G1, señalización celular), antibeta ACTINA (A1978) y antialfa TUBULINA (T9026) <p><strong>Ensayo de lactato</strong></strong>Después del tratamiento, se realizó el ensayo de L-lactato, de acuerdo con las instrucciones del fabricante. Brevemente, se tomó el sobrenadante de las células y se introdujo en la mezcla maestra de reacción. A continuación, se llevó a cabo un período de incubación de 30 minutos a temperatura ambiente en la oscuridad. Posteriormente, se midió la absorbancia a 570 nm y se calculó la cantidad de L-lactato en el medio. Además, se lisaron las células, y se examinó la cantidad total de proteínas mediante el ensayo de proteínas BCA (23227, Thermo Fisher); este paso se realizó para normalizar los valores de lactato a la cantidad de proteínas en la célula.</p> <p> </p> <p><strong>Lactato en el medio</strong> <p><strong>Radiación y ensayo clonogénico</strong></strong>Tras el tratamiento, las células se irradiaron a las dosis indicadas en un Linac de 6 MV (Varian Truebeam STx, Palo Alto, CA, EE.UU.; BrainLAB AG, Feldkirchen, Alemania) y se sembraron de nuevo en placas de 6 pocillos para la formación de colonias. Antes de la siembra, se contaron las células y se normalizaron con respecto a las condiciones de control. Tras 7-12 días, los cultivos se fijaron con violeta cristal y se contaron las colonias (>50 células). Las fracciones de supervivencia (FS) se ajustaron al modelo lineal-cuadrático utilizando el software GraphPad Prism 8 (GraphPad Prism Software Inc, San Diego, CA, EE.UU.). La radiosensibilización se evaluó en el nivel de 0,1 fracciones supervivientes.</p> <p> <p><strong>Cultivos celulares tridimensionales (3D) (esferoides)</strong></strong>Se prepararon esferoides con células EMT6 y 4T1 sembrando 4000 células/pocillo en una placa de 96 pocillos de fijación ultrabaja (Corning, Corning, NY, EE.UU.). Se añadió DCA al medio cuando los esferoides tenían alrededor de 500 µm de diámetro. A continuación, se irradiaron los esferoides con 8 Gy y se lavó el tratamiento con medio fresco que se renovaba cada 3 días. El crecimiento de los esferoides se monitorizó utilizando el IncuCyte Live Cell Imaging System (Essen Bioscience) durante 10 días.</p> <p <p><strong>Modelo tumoral en ratón<br></strong>Las células tumorales 4T1 y EMT6 (0,5 × 10<sup>6</sup>) se inocularon en la extremidad posterior izquierda de ratones Balb/c singénicos (hembra, 7-9 semanas de edad; Charles River Laboratories, L’Arbresle Cedex, Francia). Cuando los tumores alcanzaron aproximadamente 150 mm<sup>3</sup>, los ratones fueron aleatorizados y tratados durante 10 días consecutivos con DCA 300 mg/kg (intraperitoneal o intratumoral). Los ratones fueron irradiados con una dosis única de 12 Gy (tumores EMT6) o 15 Gy (tumores 4T1) o un esquema de radiación fraccionada de 5*4 Gy (tumores EMT6) y 5*6 Gy (tumores 4T1) cuando los tumores alcanzaron aproximadamente 150 mm3. La radiación se administró con un Linac de 6 MV (Varian Truebeam STx). Durante todo el transcurso del experimento, se midieron los tumores con un calibrador electrónico y se calculó el volumen tumoral mediante la fórmula Volumen = (Longitud × Anchura<sup>2</sup>) × 0,5. Los experimentos fueron aprobados por el Comité Ético para el uso de animales de laboratorio de la Vrije Universiteit Brussel (números de expediente ético: 16-552-2 (18/4/2017) y 18-552-2 (1/6/2018) </p> <p> <p><strong>Tinción con pimonidazol en secciones tumorales<br></strong>Los tumores se inocularon como se describe en la parte 4.13. Después del tratamiento, se inyectó pimonidazol (60 mg/kg; Hypoxyprobe) i.v. en la vena de la cola. Los tumores se extirparon 1,5 h más tarde, se pesaron, se congelaron y se almacenaron en viales de plástico a -80 °C. Las secciones tumorales (5 µm) se sometieron a inmunotinción utilizando un anticuerpo de conejo anti-Pimo (Hypoxyprobe, Burlington, MA, EE.UU.), que se tiñó con un anticuerpo FITC anti-conejo (Abcam). Los portaobjetos tumorales se montaron con líquido de montaje (medio de montaje DAKO, Agilent) mezclado con dapi (Sigma-Aldrich) y se cubrieron con un cubreobjetos. Las imágenes se adquirieron con microscopía confocal de fluorescencia (EVOS FL, Thermo Fisher) y se analizaron con ImageJ.</p> <p>
La citotoxicidad del DCA se evaluó mediante el ensayo MTT descrito en otro lugar <a href=»#97″>[97,</a></sup><a href=»#98″><sup>98]</sup>.</a> Brevemente, las células se cultivaron en placas de 96 pocillos y se trataron con las concentraciones indicadas. Tras el tratamiento, se aspiró el medio y se añadieron 50 µl del reactivo MTT (5 mg/mL) durante 1,5 h. A continuación, se añadieron 200 µl de disolvente MTT (19:1 DMSO/HCL) y se mezclaron para disolver los cristales de formazán generados en el interior de las células. La absorbancia se midió a una longitud de onda de 540 nm utilizando un espectrofotómetro (Bio-Rad Laboratories, Hercules, CA, EE.UU.). La viabilidad celular se determinó normalizando las células tratadas con respecto a las células de control no tratadas.</p> <p class=»spip»>Viabilidad de las células
<p><strong>Ensayo de crecimiento cinético</strong></strong>La influencia del DCA en la proliferación de las células EMT6 y 4T1 se evaluó mediante un ensayo de crecimiento cinético. Las células se cultivaron hasta confluencia en placas de cultivo de 96 pocillos y se trataron con DCA a las concentraciones indicadas en 6 repeticiones. Se tomaron microfotografías cada dos horas utilizando un generador de imágenes de células vivas Incucyte (Essen Biosciences, Newark, Reino Unido), y la confluencia de los cultivos se midió utilizando el software Incucyte (Incucyte ZOOM 2018A, Essen Biosciences) durante 80 h en cultivo.</p> <p>