Jingtao Tong, Ganfeng Xie, Jinxia He, Jianjun Li, Feng Pan y Houjie Liang
Departamento de Oncología, Hospital del Sudoeste, Tercera Universidad Médica Militar, 29 Gaotanyan Street, Chongqing 400038, China
La correspondencia debe dirigirse a Houjie Liang, [email protected]. Recibido el 27 de mayo de 2010; revisado el 29 de diciembre de 2010; aceptado el 13 de enero de 2011
Editor académico: Miguel A. Andrade
Copyright © 2011 Jingtao Tong et al. Este es un artículo de acceso abierto distribuido bajo la Licencia de Atribución Creative Commons, que permite el uso, distribución y reproducción sin restricciones en cualquier medio siempre que se cite adecuadamente el trabajo original.
Dicloroacetato (DCA), un inhibidor de la piruvato deshidrogenasa quinasa (PDK), se ha demostrado recientemente como un prometedor no tóxico agente antineoplásico que promueve la apoptosis de las células cancerosas. En el presente estudio, nos propusimos investigar el efecto antitumoral del DCA combinado con 5-fluorouracilo (5-FU) en células de cáncer colorrectal (CCR). Se trataron cuatro líneas celulares de CCR humano con DCA o 5-FU, o una combinación de DCA y 5-FU. La viabilidad celular se determinó mediante el ensayo 3-(4,5-dimetiltiazol-2-il)- 2,5-difeniltetrazolio bromuro. La interacción entre el DCA y el 5-FU se evaluó mediante el principio de effecto medio. Se realizó inmunocitoquímica con bromodesoxiuridina (BrdU) para determinar la proliferación de las células del CCR. El ciclo celular y la apoptosis se midieron mediante citometría de flujo, y la expresión de moléculas relacionadas con la apoptosis se evaluó mediante western blot. Nuestros resultados demostraron que el DCA inhibía la viabilidad de las células del CCR y presentaba una antiproliferación sinérgica en combinación con el 5-FU. Además, en comparación con el 5-FU solo, la apoptosis de las células de CCR tratadas con DCA y 5-FU se vio potenciada y demostrada con los cambios de las proteínas Bcl-2, Bax y caspasa-3. Nuestros resultados sugieren que el DCA tiene un efecto antitumoral sinérgico con el 5-FU en líneas celulares de CCR in vitro.
1. Introducción
El cáncer colorrectal es una de las neoplasias malignas más frecuentes en todo el mundo [1]. Aparte de la cirugía, el tratamiento de los pacientes con CCR se basa principalmente en la quimioterapia, especialmente en los pacientes con CCR avanzado. Entre los agentes quimioterapéuticos para el CCR, el 5-Fluorouracilo (5-FU), que es un agente quimioterapéutico clásico, ha sido el régimen de primera línea para el tratamiento del CCR durante varias décadas [2, 3]. Sin embargo, el 5-FU por sí solo es poco selectivo para el tumor, así como altamente tóxico para la médula ósea, el tracto gastrointestinal y la piel cuando se utiliza a la dosis terapéutica [4].
Las alteraciones metabólicas son una de las características más importantes del cáncer [5]. Ya en la década de 1920, Otto Warburg observó que las células cancerosas generalmente utilizan la glucólisis en lugar de la fosforilación oxidativa para obtener energía [6]. Así, el cambio metabólico a la respiración anaeróbica a través de la glucólisis a partir de piruvato, en lugar de la conversión de piruvato a acetil-CoA por la acción de la piruvato deshidrogenasa (PDH) en el metabolismo aeróbico de la glucosa, se convierte en un fenotipo preferente del progreso del cáncer. La PDH puede ser inactivada por la piruvato deshidrogenasa cinasa (PDK) en muchos fenotipos glucolíticos, incluido el cáncer, mientras que la inhibición de la PDK cambia el metabolismo a la oxidación aeróbica, lo que se ha demostrado desfavorable para el crecimiento tumoral [7].
El dicloroacetato (DCA) es un inhibidor prototípico de la PDK mitocondrial. Al bloquear la enzima, el DCA disminuye la producción de lactato al desplazar el metabolismo del piruvato de la glucólisis hacia la oxidación en la mitocondria. Esta propiedad ha llevado a ensayar el DCA para el tratamiento de trastornos de acumulación de ácido láctico [8]. Recientemente, algunos estudios han demostrado que el DCA suprime el crecimiento tumoral mediante la inhibición de la PDK [9-11]. Michelakis y sus colegas descubrieron que el DCA restablecía la función mitocondrial, restaurando así la apoptosis, matando las células cancerosas in vitro y reduciendo los tumores en las ratas [12].
La quimioterapia combinada se ha utilizado ampliamente. el 5-FU suele combinarse con otros agentes antineoplásicos y radiación para potenciar su efecto antitumoral. La insuficiencia clínica parece deberse a la resistencia del 5-FU y a sus graves efectos secundarios. La fuerte y selectiva inducción de la apoptosis sugiere que el inhibidor de la PDK DCA puede potenciar el efecto inhibidor de los fármacos contra el cáncer, superando así la eficacia del tratamiento actual. En el presente trabajo, nos propusimos examinar los efectos antitumorales combinados del DCA con el 5-FU en las células del CCR, con la esperanza de evaluar un régimen relativamente eficaz y seguro potenciado para el tratamiento del CCR.
2. Material y métodos
2.1. Células y Regentes. Las líneas celulares de cáncer de colon humano LS174T, LoVo, SW620 y HT29 se adquirieron de American Type Culture Collection (Manassas, VA, EE.UU.). Los reactivos de cultivo celular se adquirieron a Gibco-Invitrogen (Carlsbad, CA, EE.UU.). Las líneas celulares se mantuvieron en medio Eagle modificado de Dulbecco o en medio Leibovitz L-15 con un 10% de suero bovino fetal (FBS), 100 U/mL de penicilina y 100 μg/mL de estreptomicina en un incubador humidificado a 37◦C, 5% de CO2. el 5-FU y el DCA se adquirieron en Sigma-Aldrich Co. Ltd. (Louis, MO, EE.UU.), se disolvieron en agua desionizada hasta obtener una solución de trabajo de 1 mol/L, se esterilizaron por filtración y se diluyeron posteriormente en el medio de crecimiento para el tratamiento.
2.2. Ensayo de viabilidad celular. La viabilidad celular se determinó mediante el ensayo de bromuro de 3-(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazolio (MTT) (Sigma-Aldrich). Las células se sembraron en placas de 96 pocillos (5 × 104 células por pocillo) y se incubaron en condiciones de crecimiento estándar durante la noche desde el 60% hasta el 70% de confluencia. A continuación, las células se trataron con DCA solo (concentración final 0-90 mM) o con DCA combinado con 5-FU (5-200 μM). Tras un tratamiento de 48 horas, las células se incubaron a 37◦Cdurante otras 4 horas con MTT (20 μLpor pocillo) y se midió la absorbancia a 490 nm en un lector de placas BioRad Modelo 550 (Hercules, CA, EE.UU.).
2.3. Análisis de la interacción entre fármacos. La interacción entre DCA y 5-FU se analizó utilizando el principio de effect mediana descrito por Chou y Talalay [13, 14]. El programa permite calcular índices de combinación (IC) que, cuando son menores que 1, iguales a 1 o mayores que 1, indican sinergismo, aditividad o antagonismo, respectivamente, entre dos fármacos. Los IC se calcularon de la siguiente manera: (Dx)1 y (Dx)2 son las concentraciones de DCA solo o 5-FU solo, que producen un x% de inhibición del crecimiento, y (D)1 y (D)2 son las concentraciones del fármaco en combinación que inhiben el crecimiento celular también en un x%.
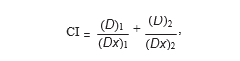
(Dx)1 y (Dx)2 se calcularon mediante la ecuación mediana-effect:
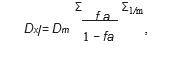
donde Dm es la dosis mediana-effect, fa es la fracción affectada, y m representa la pendiente del gráfico mediana-effect.
2.4. Ensayos de proliferación celular. La inmunocitoquímica se llevó a cabo con bromodesoxiuridina (BrdU) (BD Bioscience, San Jose, CA, EE.UU.) in vitro. Las células se propagaron sobre cubreobjetos en placas de 12 pocillos en condiciones de crecimiento estándar. Después de 24 horas, se añadieron varias concentraciones de DCA, 5-FU o una combinación de dos fármacos. Se privó a las células de suero durante 12 horas en un medio de crecimiento que contenía 0,5% de FBS para restablecer el ciclo celular a la fase G0 y, a continuación, las células recibieron un impulso durante 2 horas con 10 μmol/Lde BrdU en un medio de crecimiento. A continuación, las células se fijaron, lavaron y tiñeron siguiendo las instrucciones del fabricante.
El análisis del ciclo celular se determinó indirectamente mediante tinción con yoduro de propidio (PI, BD Bioscience) por citometría de flujo (FACScan, Becton Dickinson, San Jose, CA, EE.UU.). Las células se sembraron en placas de 6 pocillos y se cultivaron con o sin 10 mM de DCA o 20 μMde 5-FU. Tras 48 horas de incubación, las células se cosecharon con tripsina-EDTA al 0,25% (Invitrogen, Carlsbad, CA, EE.UU.). A continuación, las células se fijaron con alcohol al 70% durante 24 horas a 4◦Cy se lavaron dos veces con solución salina buffer fosfatada (PBS). Se añadió ARNasa (100 μL; 1 mg/mL) (BD Bioscience), y las células se incubaron en un baño de agua a 37◦Cdurante 30 minutos. Tras la tinción con 200 μLde PI (50 μg/mL), las células se mantuvieron a 4◦Cdurante 30 minutos. Por último, las células se analizaron por citometría de flujo.
2.5. Ensayo de apoptosis. La apoptosis se detectó mediante citometría de flujo con annexin-V-FITC (BD Bioscience) y PI. Las células se sembraron en placas de 6 pocillos. Tras una incubación de 48 horas con o sin fármacos, las células se lavaron y se resuspendieron en 0,5 mL de PBS buffer. Tras la tinción con anexina-V-FITC y PI, las células se analizaron por citometría de flujo en tres experimentos independientes.
2.6. Western Blot. Se cosecharon las células y se extrajeron las proteínas totales con buffer RIPA que contenía inhibidores de la proteasa. La proteína total (50 μg) se sometió a SDS/PAGE al 10% o al 12%, y las proteínas resueltas se transfirieron electroforéticamente a membranas de PVDF (Millipore, Bedford, MA, EE.UU.). Las membranas se bloquearon durante 2 horas con leche descremada al 5% en TBS buffer que contenía 0,05% de Tween- 20 (TBST) a 4◦C. A continuación, las membranas se incubaron con anticuerpos contra Bax, Bcl-2, Caspasa-3 y GADPH durante la noche a 4◦C. Tras lavarlas con TBST, las membranas se incubaron con sus respectivos anticuerpos secundarios durante 1 hora.
A continuación, se incubaron las membranas con el sustrato de máxima sensibilidad SuperSignal West Femto (Pierce, Rockford, IL, EE.UU.) durante 1 minuto y se tomaron imágenes con un sistema Gel Doc XR (Bio-Rad). Todos los anticuerpos se adquirieron en Santa Cruz Biotechnology (Santa Cruz, CA, EE.UU.).
2.7. 7. Estadística. Todos los datos se expresan como media de la desviación estándar (DE). El análisis estadístico se realizó con el programa SPSS 13.0 (SPSS, Chicago, IL, EE.UU.). Las diferencias entre los dos grupos se determinaron mediante la prueba t de muestras apareadas o la prueba t de muestras independientes (de dos colas), según se indica. Las diferencias entre grupos se analizaron mediante un análisis de varianza unidireccional (ANOVA). P < 0,05 se consideró estadísticamente significativo.
3. Resultados
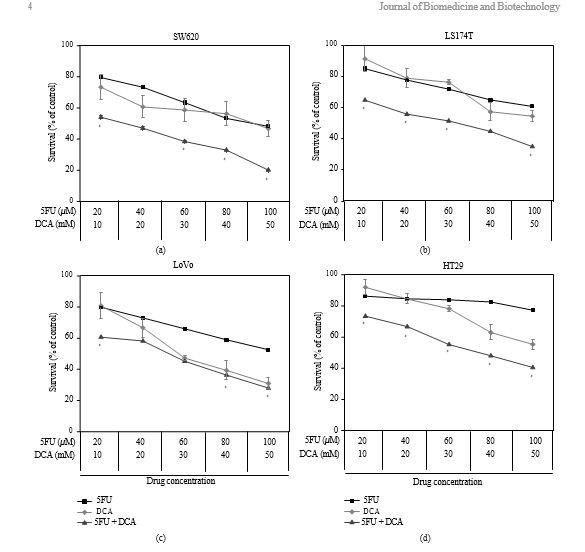
3.1. Viabilidad de las células de CCR tratadas con DCA solo o en combinación con 5-FU. Para determinar el effecto del DCA sobre las células de CCR, se expusieron las células a DCA (0-90 mM) durante 48 horas. Los resultados mostraron que el efecto inhibidor era dependiente de la dosis. Como se muestra en la figura 1, la inhibición de la viabilidad de las líneas celulares cancerosas tratadas con 50 mM de DCA fue
fue la siguiente: SW620 (46,73% ± 5,21%), LoVo (30,94% ± 3,57%), LS174t (54,59% ± 3 ,93%) y HT29 (55,31% ± 3,35%). Tratamos las células con 5-FU (20-100 μM) y descubrimos que la viabilidad de SW620, LoVo, LS174t se inhibía significativamente excepto HT29, con su inhibición de viabilidad no obvia bajo 80 μM5-FU. Cuando se trataron con estos dos fármacos simultáneamente, la viabilidad de las células de CCR mencionadas disminuyó significativamente en comparación con el DCA o el 5- FU solos (los valores IC50 se muestran en la Tabla 1).
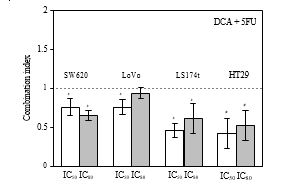
3.2. Efectos sinérgicos del DCA combinado con 5-FU en líneas celulares de CCR. La viabilidad de las células CRC disminuyó en presencia de 5-FU y DCA. El DCA potenció los efectos inhibidores del 5-FU sobre las células de CCR, y la influencia del DCA sobre los efectos del 5-FU dependió de la dosis (Figura 1). La interacción entre el 5-FU y el DCA se analizó con el método de la mediana de effectos. La combinación de DCA y 5-FU produjo efectos sinérgicos o aditivos en función del nivel de destrucción celular (Fa). Las cuatro líneas celulares de CCR mostraron efectos sinérgicos con 5-FU y DCA. El sinergismo fue estadísticamente significativo en LS174t a niveles de inhibición del 50% y el 80%, alcanzando valores de IC de 0,46-0,61, y en la línea celular HT-29 a niveles de inhibición del 50 y el 80%, con valores de IC en el intervalo de 0,42 a 0,52. En las células SW620 y LoVo, los valores de IC a niveles de inhibición del 50% y el 80% fueron de 0,64-0,75 y 0,75-0,94, respectivamente, (Figura 2).
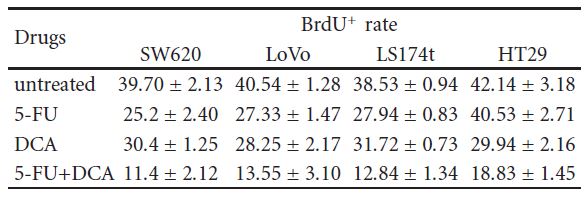
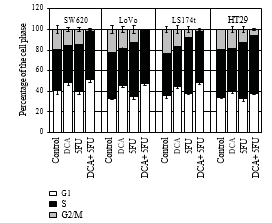
3.3. El DCA aumentó la efficiencia del efecto antiproliferativo del 5-FU. Para confirmar que la disminución de la viabilidad celular se debía a una reducción de la proliferación, se emplearon la inmunocitoquímica y la citometría de flujo. Las células se trataron con 10 mM de DCA combinado con 20 μM de 5-FU. Como era de esperar, las células tratadas con DCA mostraron una proliferación reducida en comparación con las células no tratadas. El número de células BrdU positivas en cuatro líneas celulares de CCR tras el tratamiento con 5-FU y DCA fue de 11,4 ± 2,12, 13,55 ± 3,10, 12,84 ± 1,34 y 18,83 ± 1,45, respectivamente, que fueron inferiores a las tratadas con 5-FU o DCA solos (P < 0,01, véase la Tabla 2). Además, el tratamiento con DCA potenció la detención del ciclo celular en la fase G1. Cuando se trató con DCA y 5-FU, las células bloqueadas en la fase G1/S fueron más que las incorporadas con DCA o 5-FU solos (Figura 3).
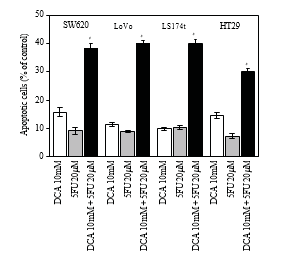
3.4. Aumento de la Apoptosis Inducida por DCA Combinado con 5-FU en Células CRC. Para investigar más a fondo la disminución de la viabilidad de las células CRC tratadas con un régimen combinado, se determinó la apoptosis mediante citometría de flujo. Como se muestra en la Figura 4, el DCA por sí solo aumentó la proporción de células CRC apoptóticas. Cuando se trataron con 10 mM de DCA, las tasas de apoptosis de cuatro líneas celulares de CCR fueron del 15,72 ± 1 ,63%, 11,32 ± 0 ,74%, 9,77 ± 0 ,53% y 14,52 ± 1 ,00%, respectivamente, mientras que las tasas de apoptosis Tabla 1: Los valores IC50 (concentraciones necesarias para reducir la viabilidad de las células en un 50% en comparación con las células de control) se calcularon mediante regresión lineal o no lineal (función de Hill de tres parámetros) (R2> 0 ,9). Se presentan como media ± DE de al menos tres experimentos independientes. Valores IC50 de los fármacos estudiados para la inhibición del crecimiento de diversas líneas celulares (las células se incubaron con los fármacos durante 48 horas).
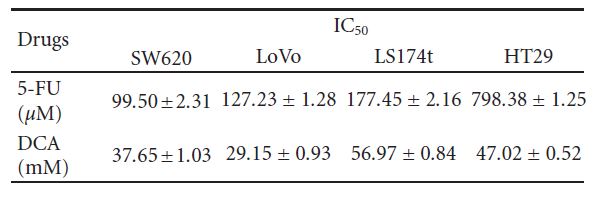
Tabla 2: Las células SW620, LoVo, LS174t, HT29 y los controles no cancerosos 293T se trataron con 10 mM de DCA y 20 μM de 5-FU solos o en combinación durante 48 horas y, a continuación, se les administró un pulso de BrdU. A continuación, se cosecharon y tiñeron las células, y el número de células BrdU+ se calculó como el número medio de células positivas en ocho campos de visión diferentes en una imagen (aumento, 400X). Este cálculo se repitió tres veces. ∗ P< 0,05, en comparación con el control. Células BrdU+ en differentes tratamientos farmacológicos (%, media ± DE).
fueron de 9,14 ± 119%, 8,82 ± 0 ,41%, 10,31 ± 0 ,71% y 7,27±0 ,96% con 5-FU. Cuando se aplicó la combinación de DCA y 5-FU, la tasa de apoptosis fue mucho mayor que con 5-FU o DCA solos (P < 0,05), lo que indicaba que el efecto apoptótico aumentaba con la combinación de DCA y 5-FU (figura 4).
3.5. Cambios en las moléculas asociadas a la apoptosis estimuladas por DCA y 5-FU. Para confirmar que el aumento de la apoptosis inducido por la terapia combinada se debía a la modificación de las expresiones de moléculas asociadas a la apoptosis, se aplicó el ensayo western blot. En la Figura 5, los resultados indicaron que el 5-FU o el DCA disminuyeron la expresión de Bcl-2 en cuatro líneas celulares de CCR en comparación con los controles de PBS, y la combinación de DCA y 5-FU disminuyó significativamente la expresión de Bcl-2 en comparación con el DCA o el 5-FU solos. Por el contrario, las expresiones de Bax y caspasa-3 aumentaron significativamente en las cuatro líneas celulares de CCR tratadas con la combinación de DCA y 5-FU en comparación con su uso único. El aumento más evidente de la expresión de Bax se detectó en las células LS174t, mientras que en LoVo parecía que la expresión de caspasa-3 era la que más aumentaba (Figura 5).
4. Discusión
En el presente estudio, demostramos que el DCA no sólo redujo la viabilidad y proliferación celular, sino que también tiene la efficiencia antitumoral sinérgica con el agente quimioterapéutico 5-FU in vitro en células de CCR.
Mientras tanto, el DCA no tiene efectos significativos en las células no cancerosas. Además, demostramos que la apoptosis inducida por el DCA contribuye a su efecto antitumoral sinérgico. En comparación con el DCA o el 5-FU por separado, el uso combinado de estos dos fármacos promueve la apoptosis de las células del CCR. el 5-FU es un fármaco quimioterápico utilizado para tratar varios tipos de cáncer, como el colorrectal, de mama, de esófago y de estómago [15]. Sin embargo, la toxicidad relacionada con el 5-FU es un problema grave y común para muchos pacientes con cáncer, siendo la mielosupresión y la toxicidad gastrointestinal los effectos secundarios más comúnmente observados [16]. La actividad clínica del 5-FU es modesta a dosis estándar y, en general, la dosificación está limitada por el perfil de seguridad. En consecuencia, solemos encontrarnos ante el dilema de tomar decisiones sobre la dosis terapéutica de 5-FU. Se han desarrollado varias estrategias para mejorar la actividad clínica del 5-FU, como la modulación bioquímica [17], las alteraciones en la programación de la administración [18] y el uso de terapias combinadas [19-22]. El DCA es una molécula pequeña, inodora, incolora, barata y relativamente no tóxica. Se utiliza clínicamente desde 1969 para el tratamiento de la acidosis láctica mediante el aumento de la capacidad de las mitocondrias para generar energía y la reducción de la acumulación de ácido láctico [23].
El DCA se ha considerado como un régimen terapéutico potencial contra el cáncer desde que un grupo canadiense descubrió que causaba regresión en varios tipos de cáncer, incluidos los de pulmón, mama y tumores cerebrales [24, 25]. Cuando se administró a células cancerosas, éstas pasaron de la glucólisis a la producción de energía mitocondrial. Es más, las mitocondrias funcionales ayudan a las células a reconocer anomalías funcionales y desencadenar la muerte celular debido a que inhiben el crecimiento tumoral e inducen la apoptosis en determinados tipos de cáncer. En el presente estudio, hallamos el mismo efecto inhibidor de la viabilidad en los CCR, que era dependiente de la dosis y variaba con los distintos grados de differenciación tumoral.
Los resultados también indicaron que la dosis baja de DCA ejerce un efecto sinérgico con el agente quimioterapéutico 5- FU en la detención del crecimiento de las células del CCR, que se analizó cuantitativamente según el método de Chou-Talalay.
Los resultados de la proliferación celular también demostraron que el DCA potenciaba los efectos antiproliferativos del 5-FU. El número de células BrdU positivas disminuyó cuando se trató con 5-FU o DCA, mientras que las células teñidas con BrdU disminuyeron significativamente cuando se trató con la combinación de 5-FU y DCA en comparación con su uso individual. Al mismo tiempo, la inhibición del crecimiento de las células del CCR se acompañó de una detención del ciclo celular. La combinación de DCA y 5-FU indujo la detención del ciclo celular en la fase G1/S en las líneas celulares de CCR, mientras que la detención inducida por 5-FU en la fase G1 no fue evidente. La inducción de la detención del ciclo celular puede ser el resultado de la inhibición de la capacidad de sintetizar o reparar el ADN, lo que puede conducir a la apoptosis celular.
El DCA parece ejercer efectos bioquímicos consistentes con la inversión del efecto Warburg y la destrucción de las células cancerosas. Descubrimos que el DCA inducía la apoptosis de las células del CCR, lo que concuerda con los estudios anteriores sobre el DCA. Es importante destacar que la combinación de DCA con 5-FU aumentó el número de células apoptóticas en comparación con 5-FU solo, lo que demuestra que el DCA inhibe el crecimiento celular a través de la apoptosis.
Muchos factores que median la apoptosis convergen para activar el effector crítico la caspasa-3, considerada como la proteasa clave de la familia de las caspasas en la apoptosis de células de mamíferos [26]. Siempre existe como precursor inactivo de 23 kD en el citoplasma, que se activa durante la apoptosis y participa en la apoptosis inducida por múltiples factores. La vía de la apoptosis dependiente de la caspasa incluye principalmente la vía mitocondrial, la vía del receptor de muerte y la vía del retículo endoplásmico [27].
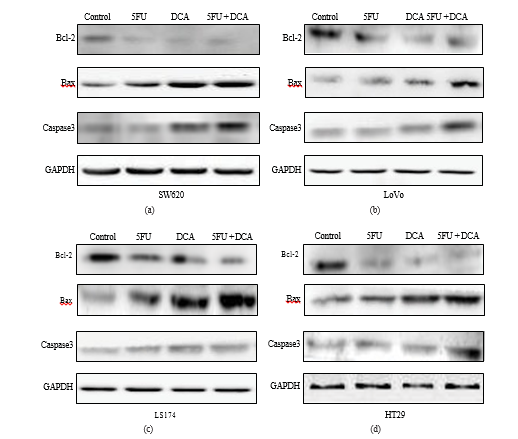
Y la vía mitocondrial está controlada y regulada por la familia de proteínas Bcl-2 [28, 29], que se dividen en dos partes, los miembros antiapoptóticos (Bcl-2) y los miembros proapoptóticos (Bax) [30]. Un estudio reciente indica que la Bcl-2 inhibe la apoptosis mediante la inhibición de la eliminación de Bax a la membrana externa mitocondrial [31]. Se investigó la expresión de caspasa-3, Bcl-2 y Bax a nivel proteico y el resultado de western blot mostró que las expresiones de caspasa-3 y Bax aumentaron, mientras que la expresión de Bcl-2 disminuyó en la combinación de DCA con el tratamiento con 5-FU, en comparación con los tratamientos individuales. Estos resultados sugieren que la apoptosis inducida por la combinación de DCA y 5-FU podría estar relacionada con la vía mitocondrial dependiente de la caspasa. Investigaciones previas sugirieron que la inducción de la apoptosis por DCA era el resultado de la disfunción de las mitocondrias y de la vía NFAT-Kv1.5 [9, 12], que se centraban en el mismo punto que el presente estudio, es decir, la apoptosis mediada por las mitocondrias.
Agradecimientos
Este trabajo ha sido financiado por la Fundación Nacional de Ciencias Naturales de China (NSFC, nº 30873015). J. Tong y G. Xie han participado a partes iguales en este trabajo.
Referencias
[1] A. Jemal, R. Siegel, E. Ward, Y. Hao, J. Xu y M. J. Thun, «Cancer statistics, 2009», CA Cancer Journal for Clinicians, vol. 59, no. 4, pp. 225-249, 2009.[2] J. A. Meyerhardt y R. J. Mayer, «Drug therapy: systemic therapy for colorectal cancer», The New England Journal of Medicine, vol. 352, no. 5, pp. 476-487, 2005.
[3] N. C. Tebbutt, E. Cattell, R. Midgley, D. Cunningham y D. Kerr, «Systemic treatment of colorectal cancer», European Journal of Cancer, vol. 38, nº 7, pp. 1000-1015, 2002.
[4] M. Gusella, A. C. Frigo, C. Bolzonella y otros, «Predictors of survival and toxicity in patients on adjuvant therapy with 5- fluorouracil for colorectal cancer», British Journal of Cancer, vol. 100, nº 10, pp. 1549-1557, 2009.
[5] D. Hanahan y R. A. Weinberg, «The hallmarks of cancer», Cell, vol. 100, nº 1, pp. 57-70, 2000.
[6] Z. Chen,W. Lu, C. Garcia-Prieto, and P.Huang, «TheWarburg effect and its cancer therapeutic implications,» Journal of Bioenergetics and Biomembranes, vol. 39, no. 3, pp. 267-274, 2007.
[7] Y. Chen, R. Cairns, I. Papandreou, A. Koong yN. C. Denko, «Oxygen consumption can regulate the growth of tumors, a new perspective on theWarburg effect», PLoS ONE, vol. 4, no. 9, Article ID e7033, 2009.
[8] A. Aynsley Green, A. M. Weindling, G. Soltesz y P. A. Jenkins, «Transient lactic acidosis and hyperalaninaemia associated with neonatal hyperinsulinaemic hypoglycaemia: the effects of dichloroacetate (DCA)», European Journal of Pediatrics, vol. 141, n.º 2, pp. 114-117, 1983.
[9] J. Y. Y. Wong,G. S. Huggins,M. Debidda, N. C.Munshi, e I. De Vivo, «Dichloroacetate induces apoptosis in endometrial cancer cells,» Gynecologic Oncology, vol. 109, no. 3, pp. 394- 402, 2008.
[10] W. Cao, S. Yacoub, K. T. Shiverick y otros, «Dichloroacetate (DCA) sensitizes both wild-type and over expressing bcl-2 prostate cancer cells in vitro to radiation», Prostate, vol. 68, nº 11, pp. 1223-1231, 2008.
[11] E. D. Michelakis, L. Webster y J. R. Mackey, «Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer», British Journal of Cancer, vol. 99, nº 7, pp. 989-994, 2008.
[12] S. Bonnet, S. L. Archer, J. Allalunis-Turner y otros, «A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth», Cancer Cell, vol. 11, nº 1, pp. 37-51, 2007.
[13] T. C. Chou y P. Talalay, «Analysis of combined drug effects: a new look at a very old problem», Trends in Pharmacological Sciences, vol. 4, pp. 450-454, 1983.
[14] T. C. Chou y P. Talalay, «Quantitative analysis of doseeffect relationships: the combined effects of multiple drugs or enzyme inhibitors,» Advances in Enzyme Regulation, vol. 22, pp. 27-55, 1984.
[15] D. B. Longley, D. P. Harkin y P. G. Johnston, «5- Fluorouracil: mechanisms of action and clinical strategies», Nature Reviews Cancer, vol. 3, no. 5, pp. 330-338, 2003.
[16] M. W. Saif, A. Choma, S. J. Salamone y E. Chu, «Pharmacokinetically guided dose adjustment of 5-fluorouracil: a rational approach to improving therapeutic outcomes», Journal of the National Cancer Institute, vol. 101, n.º 22, pp. 1543-1552, 2009.
[17] C.G. Leichman, K. Chansky, J. S. Macdonaldet al., «Biochemical modulation of 5-fluorouacil through dihydropyrimidine dehydrogenase inhibition: a Southwest OncologyGroup phase II trial of eniluracil and 5-fluorouracil in advanced resistant colorectal cancer,» Investigational New Drugs, vol. 20, no. 4, pp. 419-424, 2002.
[18] F. A. Levi, R. Zidani, J. M. Vannetzel y otros, «Chronomodulated versus fixed-infusion-rate delivery of ambulatory chemotherapy with oxaliplatin, fluorouracil, and folinic acid (leucovorin) in patients with colorectal cancer metastases: a randomized multi-institutional trial», Journal of the National Cancer Institute, vol. 86, nº 21, pp. 1608-1617, 1994.
[19] G. Melen-Mucha, E. Balcerczak, S. Mucha, M. Panczyk, S. Lipa y M. Mirowski, «Expression of p65 gene in experimental colon cancer under the influence of 5-fluorouracil given alone and in combination with hormonal modulation», Neoplasma, vol. 51, no. 4, pp. 319-324, 2004.
[20] F. Richards II, L. D. Case y D. R. White, «Combination chemotherapy (5-fluorouracil, methyl-CCNU, mitomycin C) versus 5-fluorouracil alone for advanced previously untreated colorectal carcinoma. A phase III study of the piedmont oncology association,» Journal of Clinical Oncology, vol. 4, no. 4, pp. 565-570, 1986.
[21] A. Aquino, S. P. Prete, J. W. Greiner y otros, «Effect of the combined treatment with 5-fluorouracil, γ-interferon or folinic acid on carcinoembryonic antigen expression in colon cancer cells,» Clinical Cancer Research, vol. 4, nº 10, pp. 2473- 2481, 1998.
[22] S. Obi, H. Yoshida, R. Toune y otros, «Combination therapy of intraarterial 5-fluorouracil and systemic interferon-alpha for advanced hepatocellular carcinoma with portal venous invasion», Cancer, vol. 106, nº 9, pp. 1990-1997, 2006.
[23] P. W. Stacpoole, «Review of the pharmacologic and therapeutic effects of diisopropylammonium dichloroacetate (DIPA),» The Journal of Clinical Pharmacology, vol. 9, no. 5, pp. 282- 291, 1969.
[24] S. Bonnet, S. L. Archer, J. Allalunis-Turner y otros, «A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth», Cancer Cell, vol. 11, nº 1, pp. 37-51, 2007.
[25] E. D.Michelakis, G. Sutendra, P.Dromparis et al., «Metabolic modulation of glioblastoma with dichloroacetate,» Science Translational Medicine, vol. 2, no. 31, pp. 31-ra34, 2010.
[26] T. Fernandes-Alnemri, G. Litwack, y E. S. Alnemri, «CPP32, a novel human apoptotic protein with homology to Caenorhabditis elegans cell death protein Ced-3 and mammalian interleukin-1β-converting enzyme,» The Journal of Biological Chemistry, vol. 269, no. 49, pp. 30761-30764, 1994.
[27] H. Mehmet, «Caspases find a new place to hide», Nature, vol. 403, nº 6765, pp. 29-30, 2000.
[28 ] E. Yang y S. J. Korsmeyer, «Molecular thanatopsis: a discourse on the BCL2 family and cell death», Blood, vol. 88, nº 2, pp. 386-401, 1996.
[29] D. R. Green y J. C. Reed, «Mitochondria and apoptosis,» Science, vol. 281, no. 5381, pp. 1309-1312, 1998.
[30] J. C. Reed, «Double identity for proteins of the Bcl-2 family,» Nature, vol. 387, no. 6635, pp. 773-776, 1997.
[31] B. Antonsson, S. Montessuit, B. Sanchez, y J. C. Martinou, «Bax is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells,» The Journal of Biological Chemistry, vol. 276, no. 15, pp. 11615- 11623, 2001.