Cecilie Abildgaard1, Christina Dahl1, Ahmad Abdul-Al1, Annette Christensen1 und Per Guldberg1
1 Forschungszentrum der Dänischen Krebsgesellschaft, Kopenhagen, Dänemark
Korrespondenz: Per Guldberg, E-Mail: [email protected]
Empfangen: 19 April
2017Akzeptiert: 19 July 2017Published
: 24 August 2017
Zusammenfassung
Die Dysregulation des Stoffwechsels während der Melanomprogression steht in engem Zusammenhang mit dem Erwerb genetischer und epigenetischer Veränderungen bei den Regulatoren der Stoffwechselwege. Der Retinsäurerezeptor beta (RARβ) ist in einem großen Teil der Melanome epigenetisch stillgelegt, aber eine Verbindung zwischen RARβ und der metabolischen Neuverdrahtung des Melanoms wurde noch nicht hergestellt. Hier zeigen wir, dass all-trans-Retinsäure (ein RARβ-Agonist) in primären humanen Melanozyten eine Wachstumshemmung induziert, die mit einer Abnahme des glykolytischen und oxidativen Stoffwechsels einhergeht, während eine selektive Hemmung von RARβ zu einem Anstieg der basalen glykolytischen Rate und einer erhöhten Empfindlichkeit gegenüber einer Hemmung der Glykolyse führt. In Melanomzellen förderte die Hemmung von RARβ eine geringere mitochondriale Atmung und eine höhere glykolytische Aktivität, was zu energetischem Stress und einer Aktivierung des Energiesensors AMP-aktivierte Proteinkinase führte. Diese Stoffwechselverschiebung erhöhte die Empfindlichkeit sowohl für die Hemmung der Glykolyse als auch für die Stimulation des mitochondrialen Stoffwechsels mit Dichloracetat, einem Inhibitor der Pyruvatdehydrogenase-Kinase. In Melanomzellen, die die BRAFV600E-Mutation tragen, hebt die RARβ-Aktivierung die Wirkung des BRAF-Inhibitors PLX4032 (Vemurafenib) auf. Insgesamt deuten diese Daten darauf hin, dass die RARβ-Signalübertragung an der Regulierung des zellulären Stoffwechsels beim Melanom beteiligt ist und ein potenzielles Ziel für kombinierte Behandlungsstrategien darstellen könnte.
Schlüsselwörter: Melanom, Krebsstoffwechsel, Retinsäure-Rezeptor β, mitochondriale Atmung, DichloracetatAbkürzungen
: ATRA: all-trans-Retinsäure; DCA: Dichloracetat; ECAR: extrazelluläre Versauerungsrate; OCR: Sauerstoffverbrauchsrate; ROS: reaktive Sauerstoffspezies
© Abildgaard et al. Dies ist ein Open-Access-Artikel, der unter den Bedingungen der Creative Commons Attribution License 3.0 (CC BY 3.0) verbreitet wird, die die uneingeschränkte Nutzung, Verbreitung und Vervielfältigung in jedem Medium erlaubt, sofern der ursprüngliche Autor und die Quelle genannt werden.
EINLEITUNG
Das Melanom, die tödlichste Form von Hautkrebs, verursacht jährlich 50 000 Todesfälle, wobei die Inzidenz weltweit weiter zunimmt. Während das primäre kutane Melanom durch einen chirurgischen Eingriff heilbar ist, liegt die 10-Jahres-Überlebensrate bei der am weitesten fortgeschrittenen Form der Erkrankung (Stadium IV) bei 10-15 % [1], was auf die notorische Resistenz gegen herkömmliche Krebstherapien zurückzuführen ist. Zu den jüngsten therapeutischen Fortschritten gehören Immun-Checkpoint-Inhibitoren und Therapien, die auf Onkogene oder nachgeschaltete Effektoren des MAPK-Signalwegs abzielen (z. B. BRAF- und MEK-Inhibitoren). Die Entwicklung einer erworbenen Arzneimittelresistenz führt jedoch in der Mehrzahl der Fälle zu einem Rückfall [2, 3].
Das Melanom entwickelt sich aus melaninproduzierenden Zellen, den so genannten Melanozyten, durch den Erwerb mehrerer genomischer Veränderungen. Zu den häufigsten Melanomtreibern gehören aktivierende Mutationen in BRAF und NRAS sowie inaktivierende Mutationen oder Deletionen in CDKN2A (kodiert p16INK4A und p14ARF), PTEN und TP53 [4]. Neuere Erkenntnisse deuten darauf hin, dass eine gemeinsame Funktion einiger dieser Gene in der Steuerung des Zellstoffwechsels besteht [5, 6]. Während des Fortschreitens des Melanoms wird der zelluläre Stoffwechsel umprogrammiert, was eine Verlagerung von der mitochondrialen Atmung zur aeroben Glykolyse bedeutet, was zu einem erhöhten Glukoseverbrauch und einer erhöhten Milchsäureproduktion führt (Warburg-Effekt) [7]. Mehrere Berichte auf der Grundlage von In-vitro- und In-vivo-Modellen des Melanoms und klinischen Studien mit Melanompatienten haben einen Zusammenhang zwischen aktivierenden Mutationen am Codon V600 von BRAF (am häufigsten BRAFV600E) und aerober Glykolyse nachgewiesen [8-10]. Auf molekularer Ebene reguliert BRAFV600E die oxidative Phosphorylierung durch Unterdrückung des Hauptregulators der mitochondrialen Biogenese, PGC1α, durch Hemmung des Mikrophthalmie-assoziierten Transkriptionsfaktors (MITF). Im Gegensatz dazu führt die Hemmung von BRAFV600E zu einer oxidativen Abhängigkeit durch die Induktion von PGC1α und einer erhöhten mitochondrialen Atmung [11]. Die entsprechende Abnahme der glykolytischen Aktivität lässt sich bei Melanompatienten, die mit BRAF-Inhibitoren behandelt werden, durch PET-CT-Scans sichtbar machen, die eine verringerte Aufnahme von Glukose im Tumorgewebe zeigen [10]. Klinische Phase-III-Studien mit dem BRAFV600E-Inhibitor Vemurafenib (PLX4032) zeigten eine Verbesserung des Gesamtüberlebens und des progressionsfreien Überlebens bei Patienten mit metastasiertem Melanom [12]. Mitochondrien-Inhibitoren wurden als nützliche Hilfsmittel für BRAF-Weg-Inhibitoren vorgeschlagen, um die Wirkung zu verbessern oder die Entwicklung einer Arzneimittelresistenz zu verhindern [13-15].
Zusätzlich zu den gut charakterisierten genetischen Faktoren enthält das Melanomgenom zahlreiche epigenetische Veränderungen. Eines der wiederkehrenden epigenetischen Ziele beim Melanom ist RARB, das für den Retinsäure-Rezeptor beta (RARβ) kodiert, der in 45-70 % der kutanen Melanome durch Promotor-Hypermethylierung zum Schweigen gebracht wird [16, 17]. In Zellen der melanozytären Linie vermittelt RARβ die durch Retinsäure (Vitamin A) induzierte Wachstumshemmung und Melanogenese, einem Marker der melanozytären Differenzierung [18]. Wir haben zuvor gezeigt, dass die Aktivierung von RARβ in Melanozyten eine Hochregulierung von p14ARF [17] bewirkt, das vor mitochondrialer Dysfunktion und oxidativem Stress schützt [19]. Hier zeigen wir, dass menschliche Melanozyten auf die RARβ-Aktivierung mit einer Reduzierung des oxidativen Stoffwechsels reagieren, möglicherweise als Teil einer Differenzierungsreaktion. In Melanomzellen hebt die Aktivierung von RARβ die Wirkung von PLX4032 auf, während die Hemmung von RARβ eine glykolytische Abhängigkeit und energetischen Stress induziert, was die Zellen anfällig für eine Behandlung mit dem Pyruvatdehydrogenase-Kinase-Inhibitor Dichloracetat (DCA) macht.
ERGEBNISSE
RARβ-Aktivierung reduziert das Wachstum und die Stoffwechselrate von Melanozyten
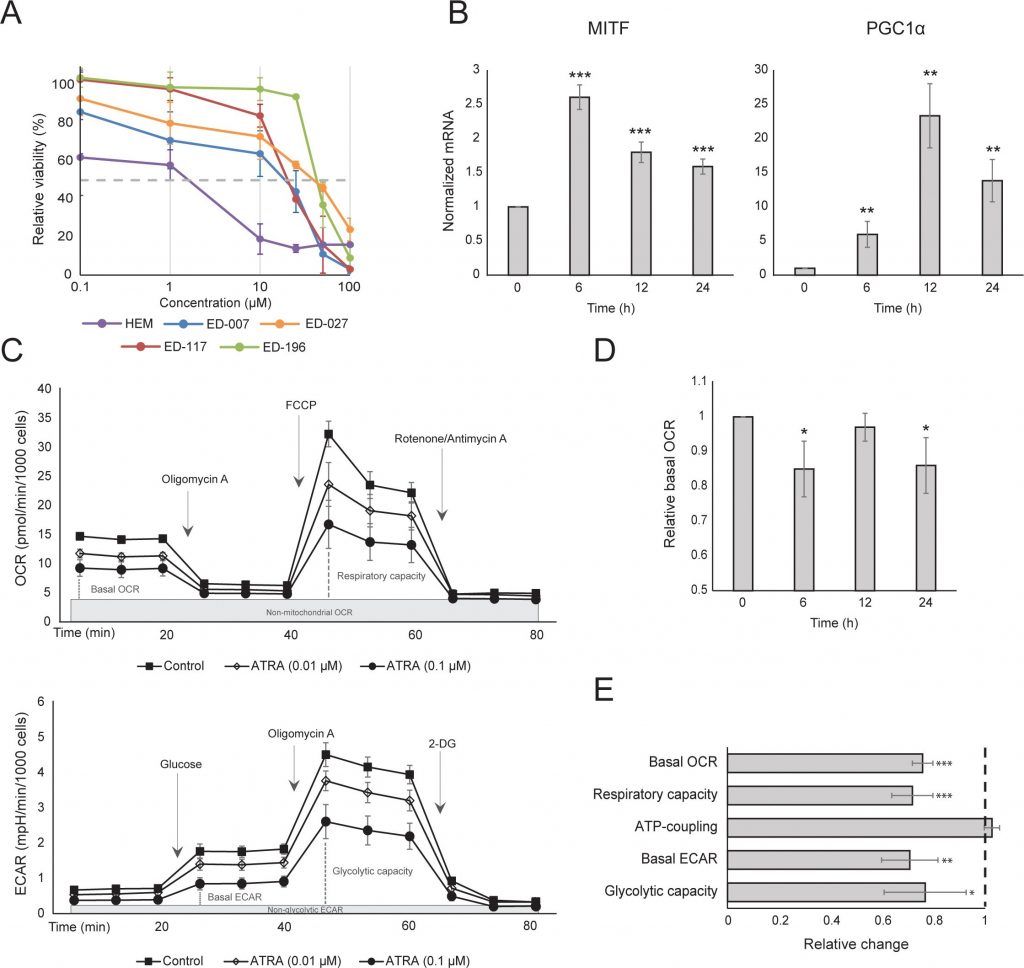
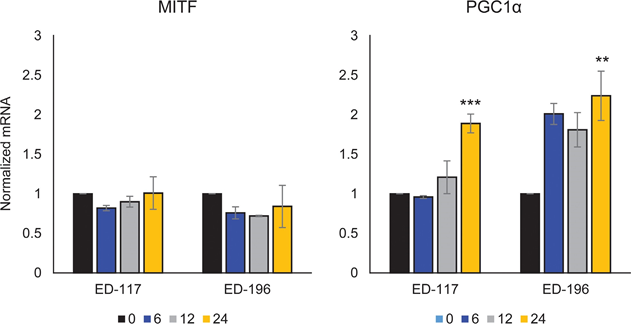
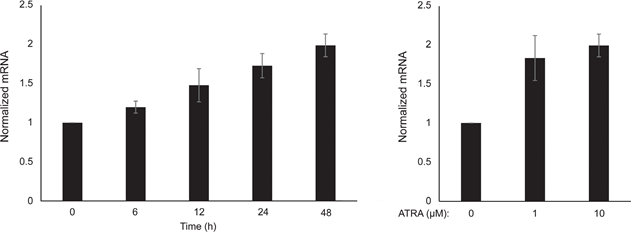
Wir haben zunächst die Wirkung der RARβ-Aktivierung auf das Wachstum von primären humanen epidermalen Melanozyten bestimmt. Die Zellen wurden 6 Tage lang mit dem RARβ-Agonisten all-trans-Retinsäure (ATRA) behandelt, und die Wachstumsreaktion wurde mit einem auf Kristallviolett basierenden Viabilitätsassay bestimmt. In Übereinstimmung mit früheren Berichten [17, 20, 21] reduzierte ATRA das Melanozytenwachstum in einer dosisabhängigen Weise (Abbildung 1A), mit einer IC50 von 2,4 μM (Tabelle 1). Es wurde bereits gezeigt, dass eine kurzfristige Behandlung (<24 h) mit ATRA die Differenzierung und Melanogenese in Melanozyten induziert, während eine langfristige Exposition (>24 h) die Proliferation reduziert und Apoptose induziert [20, 21]. Wir fanden heraus, dass ATRA (0,1 μM) eine vorübergehende Hochregulierung des für die melanozytäre Abstammungslinie spezifischen Transkriptionsfaktors MITF (Mikrophthalmie-assoziierter Transkriptionsfaktor) auslöste, wobei die Expression nach 6 Stunden ihren Höhepunkt erreichte und dann auf die Basalwerte zurückging (Abbildung 1B). In Melanomzellen reguliert MITF die Expression von PGC1α, einem Marker für einen oxidativen Phänotyp [22]. Wir untersuchten daher die Expression von PGC1α in Melanozyten zu verschiedenen Zeitpunkten nach der Exposition mit ATRA (0,1 μM). Wie in Abbildung 1B gezeigt, wurde PGC1α ebenfalls vorübergehend hochreguliert, mit einer Verzögerung von etwa 6 Stunden im Vergleich zu MITF.
| Zellen/Zelllinien | Zellen/Zelllinien | Merkmale | Merkmale | Merkmale | IC50-Werte | IC50-Werte | IC50-Werte | IC50-Werte |
|---|---|---|---|---|---|---|---|---|
| ED-Nummer | Name | BRAF-Status* | RARβ-Ausprägung** | p14ARF-Expression | ATRA (μM) | LE135 (μM) | DCA (mM)*** | PLX4032 (μM) |
| HEM# | WT | + | + | 2.4±1.6 | 2.8±0.8 | 69.1±6.4 | NA | |
| ED-007 | FM-3 | WT | + | – | 18.6±8.7 | 8.6±1.0 | 12.2±2.2 | NA |
| ED-027 | FM-82 | BRAFV600E | + | + | 39.8±5.3 | 10.7±1.3 | 17.7±2.1 | 0.52±0.04 |
| ED-117 | Mel-NT3-00 | BRAFV600E | + | + | 25.5±5.0 | NA | 37.6 ±2.2 | 0.51±0.09 |
| ED-196 | Ma-Mel-51 | BRAFV600E | + | + | 46.2±9.1 | 8.4±0.4 | 35.8±3.2 | 0.26±0.06 |
IC50-Werte sind Mittelwerte ± Standardabweichung von ≥3 unabhängigen Experimenten.
*Bestätigt durch Pyrosequenzierung**Bestätigt
durch
qPCR***IC50-Werte veröffentlicht von Abildgaard et al. [29]
#Humane epidermale Melanozyten
Aufgrund der Rolle von PGC1α bei der mitochondrialen Biogenese untersuchten wir als nächstes, ob die PGC1α-Expression mit dem Niveau der mitochondrialen Atmung korreliert. Mit dem Seahorse XFe96 Instrument haben wir die Sauerstoffverbrauchsrate (OCR) und die extrazelluläre Versauerungsrate (ECAR) gemessen, die Indikatoren für die mitochondriale Atmungsrate bzw. die glykolytische Aktivität sind. OCR und ECAR wurden während der sequentiellen Zugabe von Stoffwechselmodulatoren gemessen, so dass die Basalraten und Kapazitäten der beiden Energiesysteme bestimmt werden konnten (Abbildung 1C). Um einen Einblick in die Zeitabhängigkeit der ATRA-Reaktionen zu erhalten, wurden die Stoffwechselparameter sowohl nach kurzfristiger (6-24 h) als auch nach langfristiger (7 Tage) Exposition gemessen. Nach einer Behandlung der Melanozyten mit ATRA (0,1 μM) für 6 oder 24 Stunden war die basale OCR reduziert. Nach 12-stündiger Behandlung war die OCR jedoch ähnlich hoch wie der Ausgangswert (Abbildung 1D). Diese Schwankungen im Stoffwechselzustand fielen mit Veränderungen in der Expression von MITF und PGC1α zusammen. Eine Langzeitexposition (7 Tage) mit einer niedrigen ATRA-Dosis (0,01 μM) führte zu einer weiteren Abnahme sowohl der OCR als auch der Atmungskapazität (Abbildung 1E).
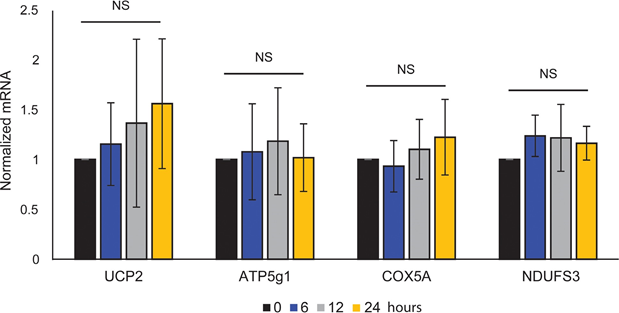
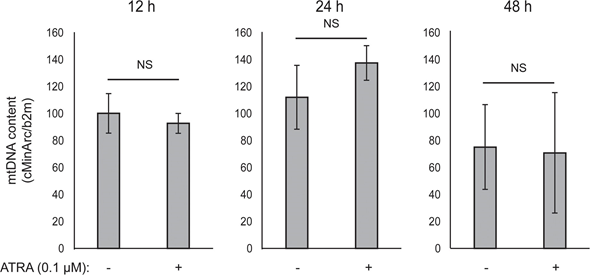
Die mitochondriale ATP-Kopplung wurde durch ATRA nicht beeinträchtigt (Abbildung 1E), was zu einer Nettoabnahme der ATP-Produktion aus den Mitochondrien führte. Es hat sich gezeigt, dass PGC1α das Entkopplungsprotein 2 (UCP2) hochreguliert, was zu einer leichten mitochondrialen Entkopplung führt [23, 24]. Die Expression von UCP2 in Melanozyten wurde durch die ATRA-Behandlung nicht beeinträchtigt (ergänzende Abbildung 1), was ein weiterer Beleg dafür ist, dass die ATP-Kopplung während der RARβ-Aktivierung unverändert blieb. Die Expression von Markern der mitochondrialen Aktivität (COX5A, ATP5g1 und NDUFS3) und der mitochondriale DNA-Gehalt wurden durch die Behandlung mit ATRA (0,1 μM) für bis zu 24 bzw. 48 Stunden ebenfalls nicht beeinträchtigt (ergänzende Abbildungen 1 und 2).
keine statistische Signifikanz (NS).
Nach 24 Stunden ATRA-Behandlung gab es keine Veränderung der glykolytischen Rate (Daten nicht gezeigt); nach 7 Tagen waren die basale glykolytische Aktivität und die glykolytische Kapazität jedoch deutlich reduziert (Abbildung 1E). Die Unterdrückung der beiden wichtigsten zellulären Energiesysteme deutet darauf hin, dass Melanozyten in Gegenwart von ATRA einen geringeren Energiebedarf aufweisen, was eine Folge des verringerten Zellwachstums sein könnte.
RARβ-Hemmung erhöht die basale glykolytische Rate und fördert die glykolytische Abhängigkeit in Melanozyten
EineHerausforderung bei der Untersuchung der zellulären Auswirkungen von ATRA ist das Vorhandensein unbekannter Konzentrationen von Vitamin A in fötalem Rinderserum, einer wesentlichen Quelle von Mikronährstoffen in den meisten Zellkulturmedien [25]. Um die Rolle der RARβ-Signalübertragung im Melanozytenstoffwechsel genauer zu untersuchen, verwendeten wir daher den RARβ-Antagonisten LE135, der mit mäßiger Selektivität gegenüber RARα und hoher Selektivität gegenüber RARγ und RXRα auf RARβ abzielt [26].
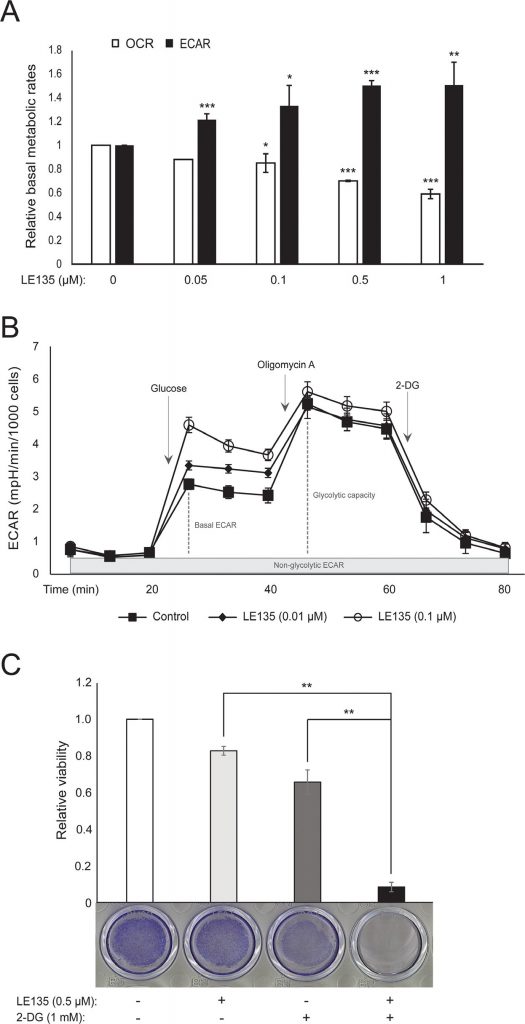
Wir wiederholten die in Abbildung 1C gezeigten Seahorse-Protokolle an Melanozyten, die 24 Stunden und 7 Tage lang mit verschiedenen Konzentrationen von LE135 behandelt wurden. Nach 24 Stunden beobachteten wir einen dosisabhängigen Anstieg der glykolytischen Aktivität, wobei die basale ECAR um bis zu 50 % anstieg, und eine entsprechende Verringerung der OCR (Abbildung 2A). Die basale ECAR war auch nach 7 Tagen der Behandlung noch erhöht (Abbildung 2B). Es gab keinen signifikanten Anstieg der glykolytischen Kapazität, was darauf hindeutet, dass die Zellen gezwungen waren, die Glykolyse zur Energiegewinnung zu nutzen. Dies wurde auch durch eine größere Empfindlichkeit dieser Zellen gegenüber dem Glykolyse-Inhibitor 2-Desoxy-D-Glukose (2-DG) in Gegenwart von LE135 bestätigt (Abbildung 2C).
ATRA hemmt die Wirkung der BRAF-Inhibition in Melanomzellen
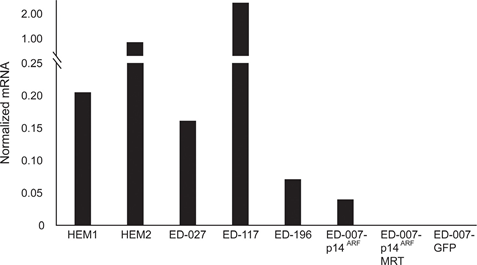
RARB wird durch Promotor-Hypermethylierung in vielen Melanomen zum Schweigen gebracht, was darauf hindeutet, dass es tumorsuppressive Eigenschaften besitzt [16]. In einer früheren Studie zu genetischen und epigenetischen Ereignissen in 110 Melanomzelllinien fanden wir eine Prävalenz von 66 % für BRAF-Mutationen und 45 % für RARB-Promotor-Hypermethylierung, wobei keine Korrelation zwischen diesen beiden Ereignissen bestand [17]. Um diese Daten zu erweitern, untersuchten wir die RARβ-Expression in 84 dieser Melanomzelllinien sowie in menschlichen epidermalen Melanozyten. Die Expressionswerte variierten stark zwischen den Melanom-Zelllinien und reichten vom völligen Fehlen der Expression bis zu Werten, die bis zu 27-mal höher waren als bei Melanozyten (Abbildung 3A). Wie erwartet war die Hypermethylierung des RARB-Promotors mit wenigen Ausnahmen mit einer geringen bis nicht nachweisbaren RARβ-Expression verbunden. Es gab keinen Zusammenhang zwischen BRAFV600E-Mutationen und RARβ-Expressionsniveaus (Abbildung 3A), was darauf hindeutet, dass die Empfindlichkeit von Melanomzellen gegenüber ATRA unabhängig vom BRAF-Status sein könnte.
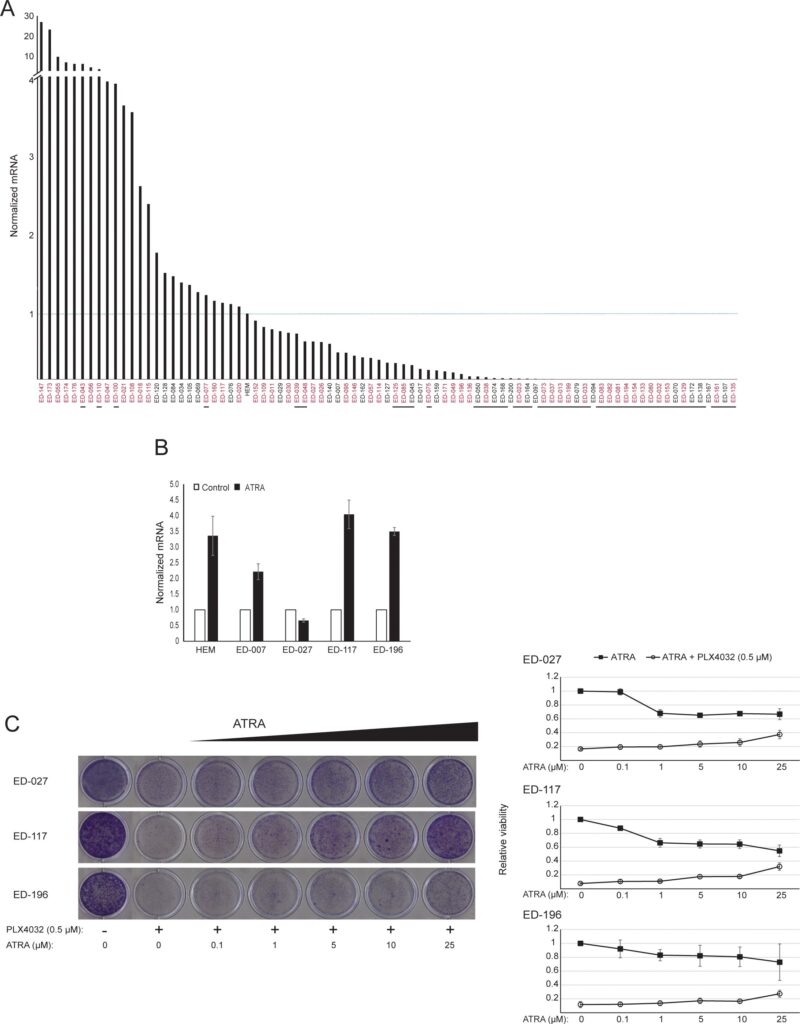
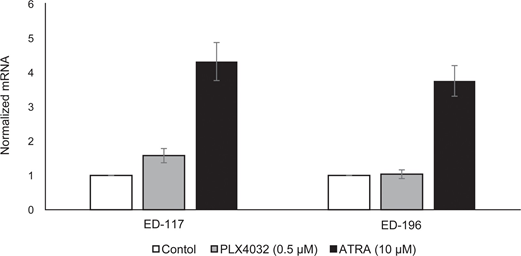
Um die Rolle der RARβ-Signalübertragung beim Melanom weiter zu untersuchen, wählten wir vier RARβ-positive Melanomzelllinien (ED-007, ED-027, ED-117 und ED-196) für eine Funktionsanalyse aus. Drei dieser Zelllinien waren BRAFV600E-mutiert (ED-027, ED-117 und ED-196) und eine war BRAF-Wildtyp (ED-007). Die Behandlung mit ATRA führte bei allen vier Zelllinien zu einem verminderten Wachstum (Abbildung 1A), obwohl ihre Empfindlichkeit geringer war als die der Melanozyten, wie aus den IC50-Werten hervorgeht (Tabelle 1). Es ist bekannt, dass die RARβ-Expression als Reaktion auf ATRA induziert wird [27]. Wie in Abbildung 3B dargestellt, wurde RARβ in 3 der vier Melanomzelllinien induziert. Trotz einer stärkeren RARβ-Induktion in ED-117 und ED-196 war die Wirkung von ATRA auf die MITF- und PGC1α-Expression im Vergleich zu Melanozyten abgeschwächt (ergänzende Abbildung 3). Die Unterdrückung der MITF/PGC1α-Achse wurde zuvor als Folge der onkogenen BRAF-Aktivität nachgewiesen [11], was zu einer geringeren Reaktion auf ATRA beitragen könnte. In Übereinstimmung mit dieser Vorstellung wiesen die BRAF-Wildtyp-Zellen die höchste Empfindlichkeit gegenüber ATRA auf, wenn auch immer noch deutlich geringer als Melanozyten (Tabelle 1).
Um zu untersuchen, ob eine gezielte Behandlung von BRAF mit PLX4032 die Empfindlichkeit gegenüber ATRA wiederherstellen würde, behandelten wir die BRAFV600E-mutierten Melanomzelllinien mit PLX4032 in einer Konzentration nahe den IC50-Werten (vgl. Tabelle 1) in Kombination mit steigenden Konzentrationen von ATRA (0,1-25 μM). Interessanterweise hob ATRA die zytotoxische Wirkung von PLX4032 in allen Zelllinien auf. Die dosisabhängige Wirkung von ATRA auf das Wachstum von Melanomzellen, die mit PLX4032 behandelt wurden (Abbildung 3C), deutet auf einen Antagonismus zwischen den beiden Verbindungen hin. Die Behandlung mit PLX4032 (0,5 μM) reduzierte die RARβ-Expression nicht (ergänzende Abbildung 4), was auf einen anderen Mechanismus für diesen Antagonismus hindeutet.
Die Wirkung von ATRA auf den Zellstoffwechsel wird durch den p14ARF-Status beeinflusst
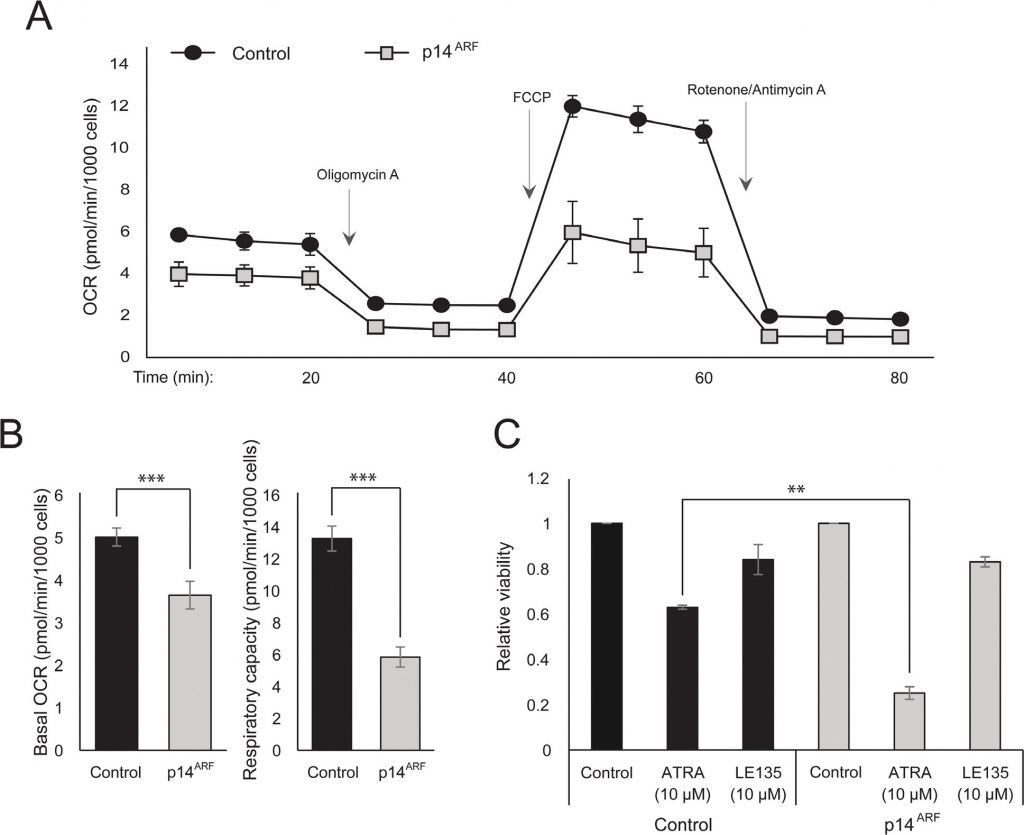
Die Beobachtung, dass sowohl ATRA als auch PLX4032 die mitochondriale Biogenese beeinflussen, könnte auf eine metabolische Erklärung für die antagonistische Wirkung dieser Verbindungen hindeuten. p14ARF wurde zuvor als zytoplasmatisches Protein in normalen Melanozyten exprimiert und schützt diese Zellen vor dysfunktionalen Mitochondrien [19]. In Übereinstimmung mit früheren Ergebnissen [17] erhöhte ATRA die Expression von p14ARF in RARβ-positiven Melanomzellen (ergänzende Abbildung 5). Obwohl p14ARF in Melanomen häufig durch Deletion des CDKN2A-Lokus verloren geht, ist es paradoxerweise nicht möglich, ARF in Melanomzellen, die dieses Gen exprimieren, stabil zu deaktivieren [17](Daten nicht gezeigt). Um stattdessen die mögliche Rolle von p14ARF bei der Vermittlung einer zellulären Reaktion auf ATRA zu untersuchen, transfizierten wir die p14ARF-defiziente Melanomzelllinie ED-007 stabil mit einem EGFP-p14ARF-Konstrukt. Die Expression von p14ARF in den transfizierten Zellen wurde mit qPCR verifiziert (ergänzende Abbildung 6). Die Seahorse-Analyse zeigte unterschiedliche Stoffwechselprofile (Abbildung 4A) mit einer signifikant niedrigeren basalen OCR und Atmungskapazität in Zellen mit wiederhergestellter p14ARF-Expression im Vergleich zu kontroll-transfizierten Zellen (Abbildung 4B). Interessanterweise zeigten die p14ARF-exprimierenden Zellen auch eine erhöhte Empfindlichkeit gegenüber ATRA (Abbildung 4C).
DieBlockierung von RARβ induziert glykolytische Abhängigkeit und energetischen Stress in Melanomzellen und sensibilisiert sie für Dichloracetat
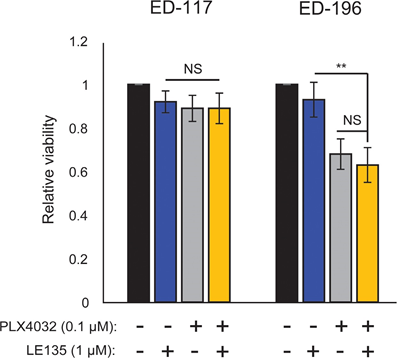
Basierend auf der Feststellung eines antagonistischen Effekts zwischen PLX4032 und ATRA testeten wir die kombinierte Wirkung von PLX4032 und LE135 auf möglichen Synergismus in ED-117 und ED-196 Melanomzellen. Bei dem hier verwendeten Versuchsaufbau (6 Tage Behandlung mit PLX4032 [0,1 μM] und LE135 [1 μM]; ergänzende Abbildung 7) wurde keine kooperative Hemmung des Melanomwachstums nachgewiesen.
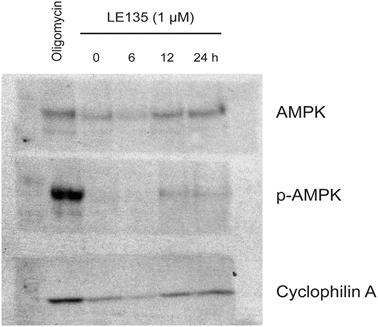
Um die Wirkung der RARβ-Hemmung auf den Melanom-Stoffwechsel weiter zu untersuchen, haben wir OCR und ECAR in RARβ-positiven Melanom-Zelllinien gemessen, die mit LE135 behandelt wurden. Ähnlich wie bei Melanozyten (Abbildung 2A-2B) erhöhte LE135 die basale glykolytische Rate und reduzierte die basale mitochondriale Atmung in Melanomzellen (Abbildung 5A-5B). Diese metabolische Verschiebung, die mit dem Warburg-Effekt übereinstimmt, war in Melanomzellen deutlicher ausgeprägt als in Melanozyten, was zu einem Anstieg der basalen glykolytischen Rate führte, um die maximale Kapazität zu erreichen. Dies wurde durch die Unfähigkeit veranschaulicht, die ECAR nach Zugabe von Oligomycin A weiter zu erhöhen, was auf einen Mangel an metabolischer Flexibilität hinweist (Abbildung 5B). Um die Wirkung von LE135 auf die zelluläre Bioenergetik zu untersuchen, untersuchten wir den Phosphorylierungsstatus der AMP-aktivierten Proteinkinase (AMPK). AMPK erkennt den zellulären Energiestatus, indem sie auf hohe AMP-Spiegel reagiert, die sich bei sinkendem ATP/ADP-Verhältnis ansammeln. So kann eine verringerte Energieproduktion oder ein erhöhter Energieverbrauch zu einem Anstieg von AMP führen, das sich an AMPK bindet und zu dessen Phosphorylierung und Aktivierung führt [28]. Die Behandlung von Melanomzelllinien mit LE135 führte zu einer Phosphorylierung von AMPK, was darauf hinweist, dass die Zellen unter energetischem Stress stehen. Der Anstieg der p-AMPK-Konzentration war bereits nach 2 Stunden zu beobachten und nahm bis mindestens 48 Stunden weiter zu (Abbildung 5C). Diese Ergebnisse unterschieden sich von denen in Melanozyten, wo die Behandlung mit LE135 nicht zu einer Aktivierung von AMPK führte (ergänzende Abbildung 8). Sowohl Melanomzellen als auch Melanozyten zeigten eine Verringerung des Zellwachstums während der 6-tägigen Behandlung mit LE135 (IC50-Werte siehe Tabelle 1). Darüber hinaus sensibilisierte LE135 Melanomzellen, ähnlich wie Melanozyten, für die glykolytische Hemmung mit 2-DG (Abbildung 5D).
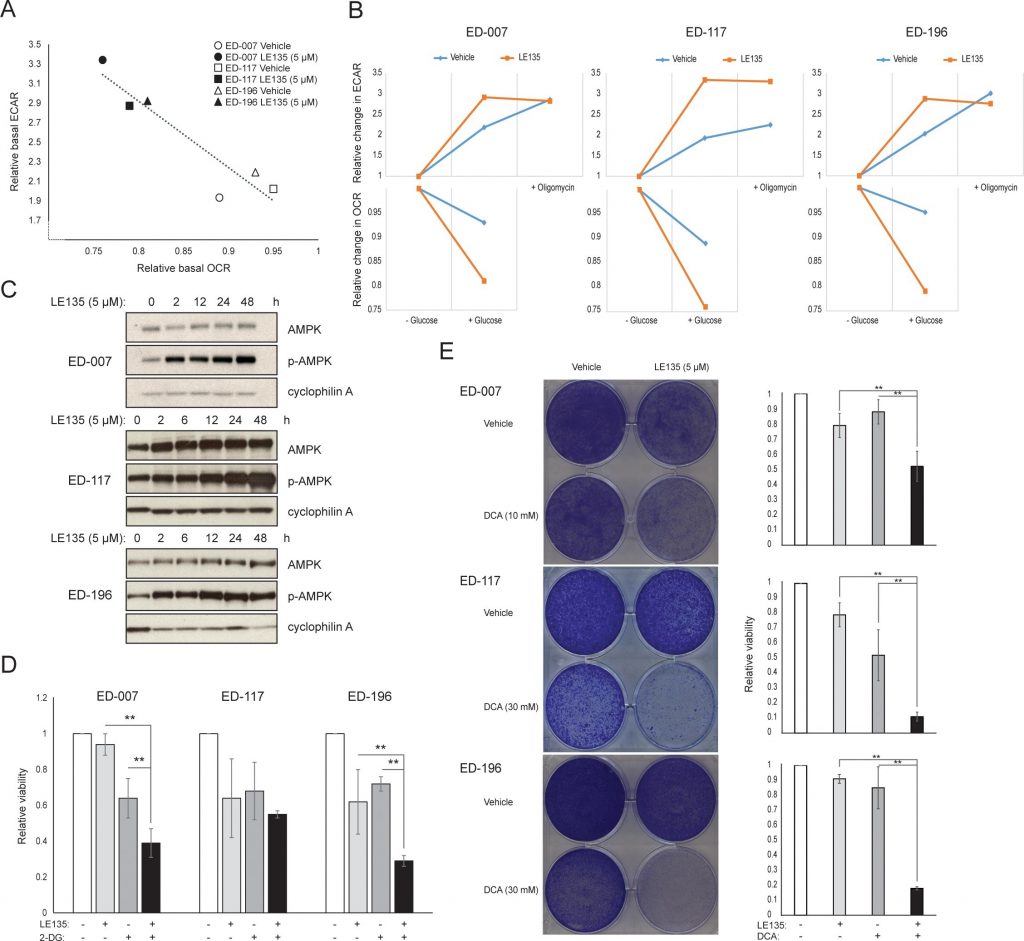
Die Induktion von energetischem Stress durch LE135 in Melanomzellen, nicht aber in Melanozyten, deutet auf eine potenzielle therapeutische Relevanz dieses Wirkstoffs in kombinierten Behandlungsstrategien hin. Der Pyruvat-Dehydrogenase-Kinase-Inhibitor DCA hat bereits gezeigt, dass er das Wachstum von Melanomzellen hemmt, indem er eine Verschiebung des Stoffwechsels weg von der Glykolyse bewirkt und die Zellen von der mitochondrialen Atmung abhängig macht [9, 29-32]. Außerdem hat sich gezeigt, dass DCA das Wachstum einer Reihe von Melanomzelllinien unabhängig vom BRAF-Status und der Empfindlichkeit gegenüber PLX4032 hemmt [29]. Um die kombinierte Wirkung von LE135 und DCA zu untersuchen, wendeten wir Konzentrationen an, die unter den jeweiligen IC50-Werten für jede der drei Melanomzelllinien lagen (vgl. Tabelle 1). Trotz der geringen Wachstumsreduktion jeder einzelnen Verbindung (9-21 % für LE135 und 12-48 % für DCA) führte die Kombination zu einer Reduktion von bis zu 89 % (Abbildung 5E). Diese Ergebnisse deuten darauf hin, dass die gegensätzlichen Wirkungen von LE135 (Förderung der glykolytischen Abhängigkeit) und DCA (Verlagerung der Zellen weg von der Glykolyse) synergistisch wirken können, um das Melanomwachstum zu hemmen.
DISKUSSION
ATRA und andere Vitamin-A-Derivate reduzieren das Zellwachstum und induzieren die Expression von Differenzierungsmarkern in verschiedenen Geweben [33, 34]. Wir fanden heraus, dass ATRA in primären menschlichen Melanozyten eine vorübergehende Hochregulierung der MITF/PGC1α-Achse bewirkt, was mit der ATRA-induzierten Steigerung der Mitochondrienfunktion übereinstimmt, die bei anderen Zelltypen wie Adipozyten und Hepatozyten beobachtet wurde [35-37]. Eine Langzeitbehandlung mit niedrigen ATRA-Konzentrationen führte jedoch zu einer Verringerung des Zellwachstums und der Stoffwechselrate, die durch eine geringere Basalglykolyse und eine geringere mitochondriale Atmung bestimmt wurde. Diese metabolischen Veränderungen als Reaktion auf ATRA spiegeln wahrscheinlich eine Differenzierungsreaktion auf den nicht-proliferativen Zustand wider, der für Melanozyten in der Haut charakteristisch ist. Die Blockierung der RARβ-Signalübertragung in diesen Zellen führte zu einem Anstieg der basalen glykolytischen Rate und einem entsprechenden Rückgang des oxidativen Stoffwechsels. Der selektive Vorteil des Verlusts der RARβ-Funktion im Melanom, zum Beispiel durch RARB-Hypermethylierung, könnte mit dem Übergang zu einem stärker glykolyseabhängigen Phänotyp zusammenhängen, der den Warburg-Effekt unterstützt.
Im Gegensatz zur Situation in primären Melanozyten führte die Blockierung der RARβ-Signalübertragung in Melanomzellen zu energetischem Stress, wie die Aktivierung von AMPK zeigt. Diese Reaktion könnte das Ergebnis einer geringeren Fähigkeit der Melanomzellen sein, die Glykolyse im Vergleich zu Melanozyten zu steigern. Während Melanozyten ein relativ niedriges glykolytisches Grundniveau haben und bei Bedarf auf eine höhere Aktivität umschalten können, zeichnen sich Melanomzellen durch eine hohe glykolytische Rate nahe ihrer maximalen Kapazität aus. Daher sind Melanozyten flexibler bei der Anpassung an die Hemmung der RARβ-Signalübertragung, um das Energieniveau aufrechtzuerhalten. Dies deutet darauf hin, dass ein relevantes therapeutisches Fenster für LE135 und andere RARβ-Inhibitoren in Kombinationstherapien, zum Beispiel mit DCA, besteht. Ähnlich wie die metabolischen Effekte der BRAF-Inhibition schaltet DCA glykolytische Krebszellen von der Glykolyse auf mitochondriale Atmung um [9, 38, 39], aber im Gegensatz zu PLX4032 ist die Wirkung von DCA nicht auf BRAF-mutierte Melanome beschränkt [29]. In einer früheren Studie haben wir gezeigt, dass die durch DCA induzierte Stoffwechselverschiebung mit einer Verringerung des ATP-Spiegels korreliert, was darauf hindeutet, dass DCA auf die bioenergetische Homöostase von Melanomzellen abzielen könnte [29]. Hier haben wir herausgefunden, dass die Kombination von LE135 und DCA das Wachstum von Melanomzellen, die den RARβ-Rezeptor exprimieren, auf kooperative Weise abschwächt. Die Behandlung von Melanomzellen entweder mit DCA oder LE135 könnte ihnen ein Zeitfenster für die Anpassung an die neuen metabolischen Anforderungen geben, während die Kombination der Behandlungen die metabolische Flexibilität einschränken und sie unfähig machen würde, die für das weitere Wachstum erforderliche Energieproduktion aufrechtzuerhalten.
Bei vielen Melanomen ist die PGC1α-Expression aufgrund der Unterdrückung von MITF durch onkogene BRAF-Mutationen gering [11]. Dieser Phänotyp unterstützt den Warburg-Effekt, indem er die Zellen zwingt, zur Energiegewinnung auf Glykolyse umzuschalten. Die Behandlung mit BRAF-Inhibitoren stellt die Expression von PGC1α wieder her und verschiebt das Stoffwechselmuster zurück zur mitochondrialen Atmung [11, 40]. Die zytotoxische Wirkung der BRAF-Hemmung kann durch das Vorhandensein von dysfunktionalen Mitochondrien in Melanomzellen verstärkt werden, was zu einer erhöhten ROS-Produktion führt [13, 41]. Wir haben festgestellt, dass ATRA das Wachstum von Melanomzellen in Gegenwart des BRAF-Inhibitors PLX4032 steigert. Auf der Grundlage der Erkenntnisse aus früheren Studien [11, 17, 19, 41] und der hier vorgestellten Ergebnisse schlagen wir ein Modell vor, das die metabolischen Wirkungen von PLX4032 und ATRA integriert und ihren Antagonismus erklärt (Abbildung 6). Das Modell deutet auf eine doppelte Rolle der verstärkten RARβ-Signalisierung hin, die zur Aktivierung von PGC1α und mitochondrialer Biogenese führt, während gleichzeitig OCR und ROS-Produktion durch Induktion von p14ARF unterdrückt werden. Das Modell wurde durch die Ergebnisse dieser und einer früheren Studie [17] gestützt, die zeigen, dass die Behandlung mit ATRA die Expression von p14ARF induziert und dass die Rekonstitution von p14ARF-defizienten Melanomzellen mit Wildtyp-p14ARF die OCR verringert und die Empfindlichkeit gegenüber ATRA erhöht. Somit könnte ATRA die Empfindlichkeit von RARβ-positiven Melanomen gegenüber PLX4032 verringern, indem es die Zytotoxizität der ROS-Produktion einschränkt. Vitamin A wurde für prophylaktische und therapeutische Zwecke bei vielen Krebsarten, einschließlich Melanomen, vorgeschlagen [42]. Obwohl die klinische Validierung noch aussteht, sprechen unsere Ergebnisse aufgrund der potenziell antagonistischen Wirkung gegen eine Vitamin-A-Supplementierung bei Melanompatienten, die mit BRAF-Inhibitoren behandelt werden.
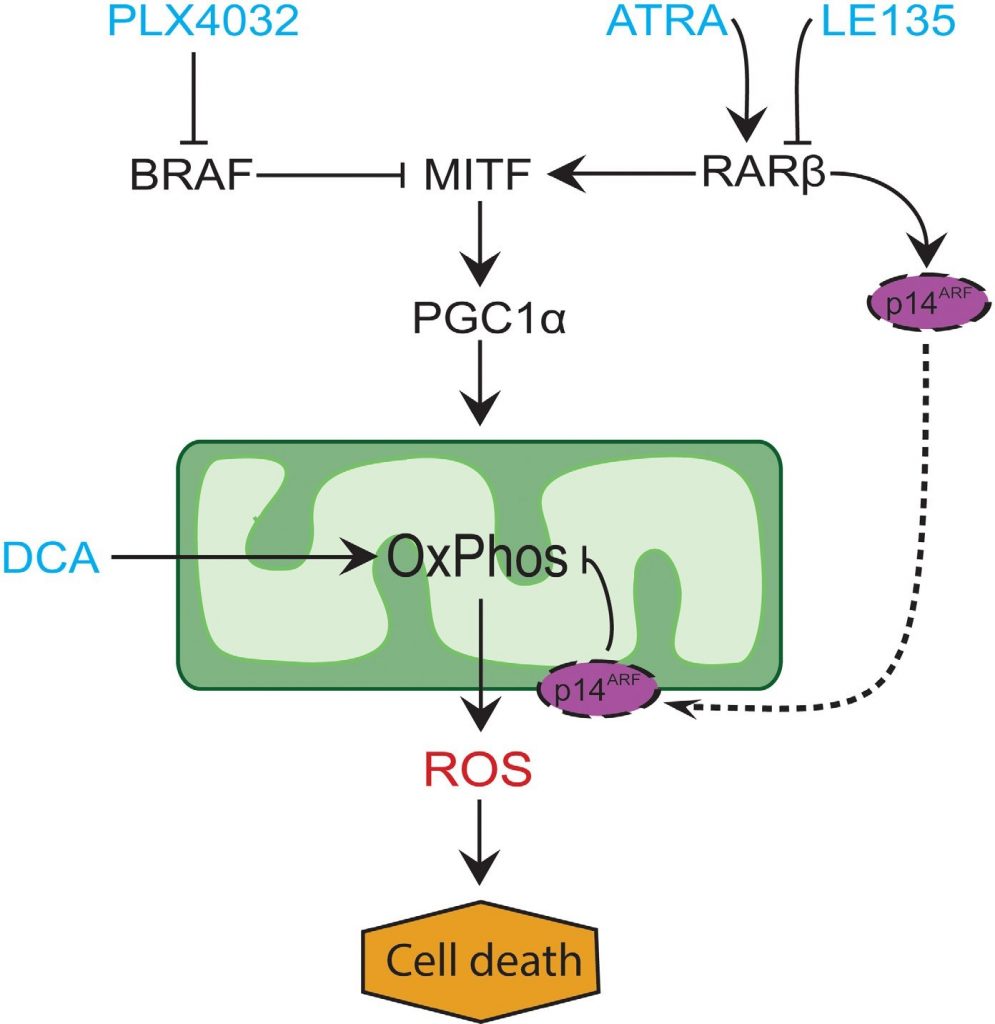
Zusammenfassend lässt sich sagen, dass wir eine neue Funktion der RARβ-Signalübertragung im Stoffwechsel von Melanozyten und Melanomzellen identifiziert haben, die klinische Auswirkungen haben könnte. Die Fähigkeit von RARβ, den MITF-PGC1α-Signalweg zu aktivieren, und eine potenziell p14ARF-abhängige Verringerung der mitochondrialen Atmungsaktivität wirken sich negativ auf das therapeutische Ansprechen auf die BRAF-Inhibition aus. Die Blockierung der RARβ-Signalübertragung fördert jedoch die glykolytische Abhängigkeit in Melanomzellen und verstärkt die Wirkung von DCA, was potenziell therapeutisch genutzt werden könnte.
MATERIALIEN UND METHODEN
Reagenzien
Natriumdichloracetat (DCA), 2-Desoxy-D-Glucose (2-DG), all-trans-Retinsäure (ATRA) und LE135 wurden von Sigma-Aldrich bezogen. DCA und 2-DG wurden in dH2O bis zu einer Arbeitskonzentration von 1 M aufgelöst. ATRA und LE135 wurden in DMSO bis zu einer Arbeitskonzentration von 0,1 M aufgelöst. PLX4032 (Vemurafenib) wurde von Selleck Chemicals erworben und in DMSO bis zu einer Arbeitskonzentration von 0,05 M aufgelöst.
Zellkultur
Melanomzelllinien wurden aus der European Searchable Tumour line Database (ESTDAB, ED) [43] bezogen. Der Status dieser Zelllinien in Bezug auf BRAF-Mutationen und RARB-Promoter-Methylierung wurde bereits beschrieben [17]. Primäre humane epidermale Melanozyten (neonatal) aus leicht pigmentiertem Gewebe (HEMn-LP; als Melanozyten bezeichnet) wurden von Invitrogen (C0025C) erworben. Für die Experimente wurden Melanozyten von drei verschiedenen Personen verwendet (Lot-Nr. 200706893 von März 2015 und November 2016; Lot-Nr. 1583282 von Februar 2017). Die Melanomzelllinien wurden bei 37 °C und 5 %CO2 in RPMI-1640-Medium kultiviert, das mit 10 % fötalem Rinderserum ergänzt wurde. HEMn-LP-Zellen wurden unter denselben Bedingungen in 254CF-Medium kultiviert, das mit 1 % humanem Melanozyten-Wachstumszusatz (HMGS) einschließlich Phorbol 12-Myristat 13-Acetat (PMA) ergänzt wurde. Für die Versuchsaufbauten wurden die HEMn-LP-Zellen in einem mit HMGS-2 (ohne PMA) ergänzten Medium kultiviert. Alle Medien und Zusätze wurden von Invitrogen bezogen.
DNA- und RNA-Reinigung
DNA für die Quantifizierung der mtDNA und RNA für die cDNA-Synthese wurden gleichzeitig mit dem AllPrep DNA/RNA/Protein mini kit (Qiagen) nach dem mitgelieferten Protokoll gereinigt.
Expressionsanalyse
Die Synthese von cDNA wurde mit dem qScript™ XLT cDNA SuperMix (Quanta Bioscience) durchgeführt. Die Genexpression von PGC1α, MITF, RARβ, p14ARF, UCP2, ATP5g1, COX5A und NDUFS3 wurde mit quantitativer Echtzeit-PCR auf dem Roche LightCycler 2.0 unter Verwendung des LightCycler FastStart DNA MasterPLUS SYBR Green I Kits (Roche) bestimmt. Die Primer sind in der ergänzenden Tabelle 1 aufgeführt.
| Gen | Vorwärts-Primer | Reverser Primer |
| PGC1α* | GTAAATCTGCGGGATGATGG | AATTGCTTGCGTCCACAAA |
| MITF** | CCGTCTCTCACTGGATTGGT | TACTTGGTGGGGTTTTCGAG |
| p14ARF*** | CCCTCGTGCTGATGCTACTGA | CATGACCTGGTCTTCTAGGAAGC |
| RARβ*** | TCCTGGATTTCTACACTGCG | AAGCAGGGTTTGTACACTCG |
| UCP2 | AAGACCATTGCCCGAGAGG | TTGGCTTTCAGGAGGGCAT |
| ATP5g1* | ATCATTGGCTATGCCAGGAA | ATGGCGAAGAGGATGAGGA |
| COX5A* | GGGAATTGCGTAAAGGGATAA | TCCTGCTTTGTCCTTAACAACC |
| NDUFS3* | GCTGACGCCCATTGAGTCTG | GGAACTCTTGGGCCAACTCC |
| RPLP0 | ACTAAAATCTCCAGGGGCACC | ATGACCAGCCCAAAGGAGAA |
qPCR*Primersequenzen veröffentlicht von Vazquez et al. 2013 [22].
**Primersequenzen veröffentlicht von Haq et al. 2014 [11].
***Primersequenzen veröffentlicht von Dahl et al. 2013 [17].
Immunoblotting
Die Proben wurden aus Zellkulturflaschen mit Lysepuffer (SLB) vorbereitet, der mit farblosem β-Mercaptoethanol (BPB), Phospho-Stop und Proteaseinhibitor (Thermo Fisher Scientific) ergänzt wurde. Die Zelllysate wurden durch Zentrifugation bei 20.000 rpm für 3 min gereinigt. Die Proteinkonzentration wurde mit dem Qubit Protein Assay Kit (Thermo Fisher Scientific) gemessen, und von jeder Probe wurden 50 μg Protein auf ein 10-Well-SDS, 4-12% Bis-Tris NuPage-Gel (Invitrogen) aufgetragen. Die Proteine wurden dann 30 Minuten lang bei 80 V und anschließend bis zum Abschluss bei 110 V getrennt. Das Blotting wurde mit einer halbtrockenen Transfereinheit auf eine ECL-Nitrocellulosemembran bei 3,3 mA/1 cm2/1 h/Gel durchgeführt. Anschließend wurde die Membran mit Ponceau angefärbt. Die Membran wurde 1 h lang in 5%iger Milch geblockt, dann zweimal 5 min lang mit TBST gewaschen und mit Anti-AMPK- oder Anti-p-AMPK (Thr172)-Antikörpern (Cell Signaling; 1:2000) in 5%iger BSA bei 4°C sowie mit einem Anti-Cyclophilin-A-Antikörper (Cell Signaling; 1:5000) als Ladekontrolle gefärbt. Nach drei 10-minütigen Waschzyklen mit TBST wurde die Membran mit dem sekundären Antikörper (Anti-Kaninchen; DakoCytomation; 1:2000) für 1 h bei Raumtemperatur gefärbt, gefolgt von drei weiteren Waschzyklen. Die Proteine wurden mit ECL Plus Western Blotting Substrate (Thermo Fisher Scientific) 1:1 für 2-3 Minuten sichtbar gemacht.
Stoffwechselanalyse
Die Stoffwechselanalyse wurde an Melanomzelllinien und Melanozyten mit einem Seahorse XFe96-Analysegerät (Seahorse Bioscience, Billerica, MA) durchgeführt, das Echtzeitmessungen der extrazellulären Versauerungsrate (ECAR) und der Sauerstoffverbrauchsrate (OCR) vornimmt. 24 Stunden vor den Messungen wurden 20 000 Zellen pro Vertiefung in Seahorse Cell Culture Microplates ausgesät. Veränderungen der Grundaktivität und der Kapazität der mitochondrialen und glykolytischen Energiesysteme wurden mit dem Mito Stress Test Kit und dem Glykolyse Stress Test Kit (Agilent Technologies) bestimmt. Die Assays wurden gemäß den mitgelieferten Protokollen durchgeführt. Der Mito-Stresstest wurde im regulären Kulturmedium durchgeführt, während beim Glykolyse-Stresstest das Medium 1 Stunde vor den Messungen durch Seahorse XF-Basismedium mit L-Glutamin (2 mM) und einem auf 7,4 eingestellten pH-Wert ersetzt wurde. Bei längeren Expositionen (>24 h) wurden die Zellen vor der Aussaat in Kulturflaschen behandelt. Alle Ergebnisse wurden auf die Anzahl der ausgesäten Zellen normalisiert, da die verwendeten ATRA- und LE135-Konzentrationen das Zellwachstum während der 24 Stunden nicht beeinflussten. Das Protokoll für die Durchführung der Assays in der Seahorse-Maschine umfasste Zyklen von 3 Minuten Mischen/3 Minuten Messen. Es wurden drei unabhängige Experimente mit 6 Wiederholungen jeder Probe durchgeführt.
Transfektion
Die Melanomzelllinie ED-007 wurde mit pEGFP (Kontrolle) oder pEGFP-p14ARF-Expressionsvektoren (2 μg Vektor/2 ×106 Zellen) transfiziert, die beide GFP als Reportergen enthalten. Die Konstrukte wurden wie zuvor beschrieben hergestellt [19]. Die Transfektion erfolgte mit der Amaxa-Nukleofektionstechnologie, Puffer V, Programm T-020, gemäß dem vom Hersteller empfohlenen Protokoll. Die erfolgreiche Transfektion wurde visuell überprüft. Stabile Klone wurden mit 400 μg/ml G418 (Genetin; Thermo Fisher Scientific) selektiert. Bei der Versuchsanordnung wurden die Zellen ohne G418 ausgesät.
Quantifizierung der mitochondrialen DNA
Die Quantifizierung der mitochondrialen DNA erfolgte mittels digitaler Tröpfchen-Polymerase-Kettenreaktion (ddPCR) unter Verwendung des QX200-Systems (BioRad Laboratories, Hercules, CA, USA). Für jede Reaktion wurden ca. 0,5 ng DNA verwendet. Die mitochondriale Kopienzahl wurde durch Berechnung des Verhältnisses zwischen einer mitochondrialen DNA-Stelle (mtMinArc) und einem Kernlocus mit einer Kopie (β2m) bestimmt, wie von Phillips et al. beschrieben [44]. Primer, Sonden und experimentelle Bedingungen sind in der ergänzenden Tabelle 2 aufgeführt.
| mtMinArc | β2m | |
| Vorwärtsprimer* | CTAAATAGCCCACACGTTCCC | GCTGGGTAGCTCTAAACAATGTATTCA |
| Umgekehrter Primer* | AGAGCTCCCGTGAGTGGTTA | CCATGTACTAACAAATGTCTAAAATGGT |
| Sonde* | 6FAM-CATCACGATGGATCACAGGT(NFQ) | VIC-CAGCAGCCTATTCTGC(NFQ) |
| Primer-Konz. | 75 nM | 500 nM |
| Glühtemp. | 50°C | 52°C |
| Anzahl der Zyklen | 40 | 40 |
und Sondensequenzen, die von Phillips et al. veröffentlicht wurden [44].
Statistische Analyse
Unterschiede zwischen unabhängigen Datensätzen wurden mit dem Student’s t-Test ermittelt. Für die statistische Analyse der Varianz zwischen verschiedenen Behandlungen wurde eine einseitige ANOVA mit übereinstimmenden Stichproben verwendet. Zur Bestimmung der statistischen Signifikanz wurde der Tukey’s honest significance difference (HSD) Multivergleichstest verwendet.
Beiträge der Autoren
CA und PG planten und organisierten die Studie. CA führte den Großteil der Experimente und die Verarbeitung der Daten durch, einschließlich Zellkultur, Behandlungsprotokolle, Stoffwechselanalyse und Statistik. CD plante und führte die EGFP-p14ARF-Transfektion und mtDNA-Messungen durch und half bei der Interpretation der Ergebnisse. AA führte Immunoblotting- und quantitative PCR-Protokolle durch und optimierte sie. AC führte Zellkulturuntersuchungen durch. CA und PG verfassten das Manuskript mit Beiträgen und Korrekturen von CD, AA und AC. Das endgültige Manuskript wurde von allen Autoren gelesen und genehmigt.
INTERESSENKONFLIKTE
Die Autoren erklären, dass sie keine Interessenkonflikte haben.
FÖRDERUNG
Diese Studie wurde von der Dänischen Krebsgesellschaft unterstützt
REFERENZEN
1 Überlebensraten für Melanom-Hautkrebs, nach Stadium. (cancer.org: Amerikanische Krebsgesellschaft).2 Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, Chodon T, Nelson SF, McArthur G, et al. Melanome erwerben Resistenz gegen B-RAF (V600E)-Hemmung durch RTK- oder N-RAS-Hochregulierung. Nature. 2010; 468: 973-7. https://doi.org/10.1038/nature09626.
3 Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP, Barretina J, Caponigro G, Hieronymus H, Murray RR, et al. COT treibt die Resistenz gegen RAF-Inhibition durch Reaktivierung des MAP-Kinase-Wegs an. Nature. 2010; 468: 968-72. https://doi.org/10.1038/nature09627.
4 Miller AJ, Mihm MC Jr. Melanom. N Engl J Med. 2006; 355: 51-65. https://doi.org/10.1056/NEJMra052166.
5 Abildgaard C, Guldberg P. Molecular drivers of cellular metabolic reprogramming in melanoma. Trends Mol Med. 2015; 21: 164-71. https://doi.org/10.1016/j.molmed.2014.12.007.
6 Ratnikov BI, Scott DA, Osterman AL, Smith JW, Ronai ZA. Metabolic rewiring in melanoma. Oncogene. 2017; 36: 147-57. https://doi.org/10.1038/onc.2016.198.
7 Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927; 8: 519-30.
8 Hall A, Meyle KD, Lange MK, Klima M, Sanderhoff M, Dahl C, Abildgaard C, Thorup K, Moghimi SM, Jensen PB, Bartek J, Guldberg P, Christensen C. Dysfunktionale oxidative Phosphorylierung macht maligne Melanomzellen süchtig nach Glykolyse, angetrieben durch das (V600E) BRAF-Onkogen. Oncotarget. 2013; 4: 584-99. https://doi.org/10.18632/oncotarget.965.
9 Parmenter TJ, Kleinschmidt M, Kinross KM, Bond ST, Li J, Kaadige MR, Rao A, Sheppard KE, Hugo W, Pupo GM, Pearson RB, McGee SL, Long GV, et al. Response of BRAF-mutant melanoma to BRAF inhibition is mediated by a network of transcriptional regulators of glycolysis. Cancer Discov. 2014; 4: 423-33. https://doi.org/10.1158/2159-8290.CD-13-0440.
10 McArthur GA, Puzanov I, Amaravadi R, Ribas A, Chapman P, Kim KB, Sosman JA, Lee RJ, Nolop K, Flaherty KT, Callahan J, Hicks RJ. Ausgeprägtes, homogenes und frühes [18F]Fluordesoxyglukose-Positronenemissionstomographie-Ansprechen auf Vemurafenib bei BRAF-mutiertem fortgeschrittenem Melanom. J Clin Oncol. 2012; 30: 1628-34. https://doi.org/10.1200/JCO.2011.39.1938.
11 Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, Frederick DT, Hurley AD, Nellore A, Kung AL, Wargo JA, Song JS, Fisher DE, et al. Oncogenic BRAF reguliert den oxidativen Stoffwechsel über PGC1alpha und MITF. Cancer Cell. 2013; 23: 302-15. https://doi.org/10.1016/j.ccr.2013.02.003.
12 Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011; 364: 2507-16. https://doi.org/10.1056/NEJMoa1103782.
13 Corazao-Rozas P, Guerreschi P, Jendoubi M, Andre F, Jonneaux A, Scalbert C, Garcon G, Malet-Martino M, Balayssac S, Rocchi S, Savina A, Formstecher P, Mortier L, et al. Mitochondrial oxidative stress is the Achille’s heel of melanoma cells resistant to Braf-mutant inhibitor. Oncotarget. 2013; 4: 1986-98. https://doi.org/10.18632/oncotarget.1420.
14 Zhang G, Frederick DT, Wu L, Wei Z, Krepler C, Srinivasan S, Chae YC, Xu X, Choi H, Dimwamwa E, Ope O, Shannan B, Basu D, et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J Clin Invest. 2016; 126: 1834-56. https://doi.org/10.1172/JCI82661.
15 Livingstone E, Swann S, Lilla C, Schadendorf D, Roesch A. Combining BRAF(V) (600E) inhibition with modulators of the mitochondrial bioenergy metabolism to overcome drug resistance in metastatic melanoma. Exp Dermatol. 2015; 24: 709-10. https://doi.org/10.1111/exd.12718.
16 Hoon DS, Spugnardi M, Kuo C, Huang SK, Morton DL, Taback B. Profiling epigenetic inactivation of tumor suppressor genes in tumors and plasma from cutaneous melanoma patients. Oncogene. 2004; 23: 4014-22. https://doi.org/10.1038/sj.onc.1207505.
17 Dahl C, Christensen C, Jonsson G, Lorentzen A, Skjodt ML, Borg A, Pawelec G, Guldberg P. Mutual exclusivity analysis of genetic and epigenetic drivers in melanoma identifies a link between p14 ARF and RARbeta signaling. Mol Cancer Res. 2013; 11: 1166-78. https://doi.org/10.1158/1541-7786.MCR-13-0006.
18 Lotan R, Lotan D. Enhancement of melanotic expression in cultured mouse melanoma cells by retinoids. J Cell Physiol. 1981; 106: 179-89. https://doi.org/10.1002/jcp.1041060203.
19 Christensen C, Bartkova J, Mistrik M, Hall A, Lange MK, Ralfkiaer U, Bartek J, Guldberg P. A short acidic motif in ARF guards against mitochondrial dysfunction and melanoma susceptibility. Nat Commun. 2014; 5: 5348. https://doi.org/10.1038/ncomms6348.
20 Baldea I, Costin GE, Shellman Y, Kechris K, Olteanu ED, Filip A, Cosgarea MR, Norris DA, Birlea SA. Biphasic pro-melanogenic and pro-apoptotic effects of all-trans-retinoic acid (ATRA) on human melanocytes: time-course study. J Dermatol Sci. 2013; 72: 168-76. https://doi.org/10.1016/j.jdermsci.2013.06.004.
21 Kawakami T, Ohgushi A, Hirobe T, Soma Y. Analysis of the effects of all-trans retinoic acid on human melanocytes and melanoblasts in vitro. J Dermatol. 2017; 44: 93-4. https://doi.org/10.1111/1346-8138.13477.
22 Vazquez F, Lim JH, Chim H, Bhalla K, Girnun G, Pierce K, Clish CB, Granter SR, Widlund HR, Spiegelman BM, Puigserver P. PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell. 2013; 23: 287-301. https://doi.org/10.1016/j.ccr.2012.11.020.
23 Donadelli M, Dando I, Fiorini C, Palmieri M. UCP2, a mitochondrial protein regulated at multiple levels. Cell Mol Life Sci. 2014; 71: 1171-90. https://doi.org/10.1007/s00018-013-1407-0.
24 Oberkofler H, Klein K, Felder TK, Krempler F, Patsch W. Role of peroxisome proliferator-activated receptor-gamma coactivator-1alpha in the transcriptional regulation of the human uncoupling protein 2 gene in INS-1E cells. Endokrinologie. 2006; 147: 966-76. https://doi.org/10.1210/en.2005-0817.
25 Arigony AL, de Oliveira IM, Machado M, Bordin DL, Bergter L, Pra D, Henriques JA. Der Einfluss von Mikronährstoffen in der Zellkultur: eine Betrachtung der Lebensfähigkeit und der genomischen Stabilität. Biomed Res Int. 2013; 2013: 597282. https://doi.org/10.1155/2013/597282.
26 Li Y, Hashimoto Y, Agadir A, Kagechika H, Zhang X. Identification of a novel class of retinoic acid receptor beta-selective retinoid antagonists and their inhibitory effects on AP-1 activity and retinoic acid-induced apoptosis in human breast cancer cells. J Biol Chem. 1999; 274: 15360-6.
27 de The H, Marchio A, Tiollais P, Dejean A. Differential expression and ligand regulation of the retinoic acid receptor alpha and beta genes. EMBO J. 1989; 8: 429-33.
28 Hardie DG, Hawley SA. AMP-aktivierte Proteinkinase: die Energieladungshypothese revisited. Bioessays. 2001; 23: 1112-9. https://doi.org/10.1002/bies.10009.
29 potenziert ihre Reaktion auf die Hemmung von BRAFV600E. J Transl Med. 2014; 12: 247. https://doi.org/10.1186/s12967-014-0247-5.
30 Populo H, Caldas R, Lopes JM, Pardal J, Maximo V, Soares P. Overexpression of pyruvate dehydrogenase kinase supports dichloroacetate as a candidate for cutaneous melanoma therapy. Expert Opin Ther Targets. 2015; 19: 733-45. https://doi.org/10.1517/14728222.2015.1045416.
31 Kluza J, Corazao-Rozas P, Touil Y, Jendoubi M, Maire C, Guerreschi P, Jonneaux A, Ballot C, Balayssac S, Valable S, Corroyer-Dulmont A, Bernaudin M, Malet-Martino M, et al. Inactivation of the HIF-1alpha/PDK3 signaling axis drives melanoma towards mitochondrial oxidative metabolism and potentiates the therapeutic activity of pro-oxidants. Cancer Res. 2012; 72: 5035-47. https://doi.org/10.1158/0008-5472.CAN-12-0979.
32 Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, van der Burg SH, Verdegaal EM, Cascante M, Shlomi T, Gottlieb E, Peeper DS. Eine Schlüsselrolle für die mitochondriale Gatekeeper-Pyruvatdehydrogenase bei onkogen-induzierter Seneszenz. Nature. 2013; 498: 109-12. https://doi.org/10.1038/nature12154.
33 Murholm M, Isidor MS, Basse AL, Winther S, Sorensen C, Skovgaard-Petersen J, Nielsen MM, Hansen AS, Quistorff B, Hansen JB. Retinsäure hat unterschiedliche Auswirkungen auf die UCP1-Expression in Maus und menschlichen Adipozyten. BMC Cell Biol. 2013; 14: 41. https://doi.org/10.1186/1471-2121-14-41.
34 Jin W, Xu YP, Yang AH, Xing YQ. In vitro-Induktion und Differenzierung von mesenchymalen Stammzellen aus der Nabelschnur in neuronenähnliche Zellen durch all-trans-Retinsäure. Int J Ophthalmol. 2015; 8: 250-6. https://doi.org/10.3980/j.issn.2222-3959.2015.02.07.
35 Tourniaire F, Musinovic H, Gouranton E, Astier J, Marcotorchino J, Arreguin A, Bernot D, Palou A, Bonet ML, Ribot J, Landrier JF. All-trans-Retinsäure induziert die oxidative Phosphorylierung und Mitochondrien-Biogenese in Adipozyten. J Lipid Res. 2015; 56: 1100-9. https://doi.org/10.1194/jlr.M053652.
36 Tripathy S, Chapman JD, Han CY, Hogarth CA, Arnold SL, Onken J, Kent T, Goodlett DR, Isoherranen N. All-trans-Retinsäure verbessert die mitochondriale Funktion in Modellen der menschlichen Leber. Mol Pharmacol. 2016; 89: 560-74. https://doi.org/10.1124/mol.116.103697.
37 Watabe H, Soma Y, Ito M, Kawa Y, Mizoguchi M. All-trans-Retinsäure induziert die Differenzierung und Apoptose von murinen Melanozytenvorläufern durch Induktion des Mikrophthalmie-assoziierten Transkriptionsfaktors. J Invest Dermatol. 2002; 118: 35-42. https://doi.org/10.1046/j.0022-202x.2001.01614.x.
38 De Preter G, Neveu MA, Danhier P, Brisson L, Payen VL, Porporato PE, Jordan BF, Sonveaux P, Gallez B. Inhibition of the pentose phosphate pathway by dichloroacetate unravels a missing link between aerobic glycolysis and cancer cell proliferation. Oncotarget. 2016; 7: 2910-20. https://doi.org/10.18632/oncotarget.6272.
39 Michelakis ED, Webster L, Mackey JR. Dichloracetat (DCA) als potenzielle metabolische zielgerichtete Therapie für Krebs. Br J Cancer. 2008; 99: 989-94. https://doi.org/10.1038/sj.bjc.6604554.
40 Corazao-Rozas P, Guerreschi P, Andre F, Gabert PE, Lancel S, Dekiouk S, Fontaine D, Tardivel M, Savina A, Quesnel B, Mortier L, Marchetti P, Kluza J. Mitochondrial oxidative phosphorylation controls cancer cell’s life and death decisions upon exposure to MAPK inhibitors. Oncotarget. 2016; 7: 39473-85. https://doi.org/10.18632/oncotarget.7790.
41 Bauer D, Werth F, Nguyen HA, Kiecker F, Eberle J. Critical role of reactive oxygen species (ROS) for synergistic enhancement of apoptosis by vemurafenib and the potassium channel inhibitor TRAM-34 in melanoma cells. Cell Death Dis. 2017; 8: e2594. https://doi.org/10.1038/cddis.2017.6.
42 Chen MC, Hsu SL, Lin H, Yang TY. Retinoic acid and cancer treatment. Biomedicine (Taipei). 2014; 4: 22. https://doi.org/10.7603/s40681-014-0022-1.
43 Robinson J, Roberts CH, Dodi IA, Madrigal JA, Pawelec G, Wedel L, Marsh SG. The European searchable tumour line database. Cancer Immunol Immunother. 2009; 58: 1501-6. https://doi.org/10.1007/s00262-008-0656-5.
44 Phillips NR, Sprouse ML, Roby RK. Simultane Quantifizierung der mitochondrialen DNA-Kopienzahl und des Deletionsverhältnisses: ein Multiplex-Echtzeit-PCR-Assay. Sci Rep. 2014; 4: 3887. https://doi.org/10.1038/srep03887.