Sven de Mey 1, Inès Dufait 1, Heng Jiang 1, Cyril Corbet 2, Hui Wang 1, Melissa Van De Gucht 1, Lisa Kerkhove 1, Ka Lun Law 1, Hugo Vandenplas 3, Thierry Gevaert 1, Olivier Feron 2 und Mark De Ridder 1,*
1 Abteilung für Strahlentherapie, Universitair Ziekenhuis Brussel, Vrije Universiteit Brussel, 1090 Brüssel, Belgien; [email protected] (S.d.M.); [email protected] (I.D.); [email protected] (H.J.); [email protected] (H.W.); [email protected] (M.V.D.G.); [email protected] (L.K.); [email protected] (K.L.L.); [email protected] (T.G.))
2 Pole of Pharmacology and Therapeutics (FATH), Institut de Recherche Expérimentale et Clinique (IREC), UCLouvain, 1200 Brüssel, Belgien; [email protected] (C.C.); [email protected] (O.F.)
3 Department of Medical Oncology, Universitair Ziekenhuis Brussel, Vrije Universiteit Brussel, 1090 Brüssel, Belgien;
[email protected]:
[email protected]: 14 September 2020Accepted
: 4. Dezember 2020Veröffentlicht
: 9. Dezember 2020
Zusammenfassung
Der mitochondriale Stoffwechsel ist ein attraktives Ziel für die Krebstherapie. Die Umprogrammierung von Stoffwechselwegen kann potenziell Tumoren mit begrenzten Behandlungsmöglichkeiten, wie dreifach negativer Brustkrebs (TNBC), für Chemo- und/oder Strahlentherapie sensibilisieren. Dichloracetat (DCA) ist ein spezifischer Inhibitor der Pyruvat-Dehydrogenase-Kinase (PDK), was zu einer erhöhten Produktion reaktiver Sauerstoffspezies (ROS) führt. ROS sind die primären Effektormoleküle der Strahlung, und eine Zunahme dieser Spezies verstärkt die Strahlenreaktion. In dieser Studie haben wir die Auswirkungen von DCA und Strahlentherapie auf zwei TNBC-Zelllinien, nämlich EMT6 und 4T1, unter aeroben und hypoxischen Bedingungen untersucht. Wie erwartet, verringerte die DCA-Behandlung die phosphorylierte Pyruvatdehydrogenase (PDH) und senkte sowohl die extrazelluläre Azidifikationsrate (ECAR) als auch die Laktatproduktion. Bemerkenswerterweise führte die DCA-Behandlung zu einem signifikanten Anstieg der ROS-Produktion (bis zum 15-Fachen) in hypoxischen Krebszellen, nicht aber in aeroben Zellen. Folgerichtig bewirkte DCA eine Radiosensibilisierung von hypoxischen Tumorzellen und 3D-Sphäroiden, während die intrinsische Radiosensitivität der Tumorzellen unverändert blieb. Unsere Ergebnisse deuten darauf hin, dass DCA, obwohl es als ein die oxidative Phosphorylierung (OXPHOS) förderndes Medikament beschrieben wird, auch die hypoxische Radiosensitivität erhöhen kann. Diese Studie ebnet daher den Weg für die gezielte Beeinflussung des mitochondrialen Stoffwechsels von hypoxischen Krebszellen, insbesondere zur Bekämpfung der Radioresistenz.
Schlüsselwörter: Dichloracetat; hypoxische Radiosensitivität; Brustkrebs; reaktive Sauerstoffspezies
© 2020 by the authors. Lizenznehmer MDPI, Basel, Schweiz. Dieser Artikel ist ein Open-Access-Artikel, der unter den Bedingungen der Creative-Commons-Attribution (CC BY)-Lizenz verbreitet wird (http://creativecommons.org/licenses/by/4.0/).
EINLEITUNG
Brustkrebs ist weltweit die häufigste Krebserkrankung bei Frauen und führt jährlich zu 627.000 Todesfällen [1]. In den letzten Jahrzehnten wurden erhebliche Fortschritte bei der Behandlung von Brustkrebs erzielt. Für Patientinnen mit dreifach-negativem/basalem Brustkrebs stehen jedoch nur begrenzte Therapien zur Verfügung [2,3,4]. Die Standardtherapie für die Behandlung von Hochrisiko-Brustkrebs besteht aus einer neoadjuvanten Chemotherapie und einer Operation, gefolgt von einer postoperativen Bestrahlung der gesamten Brust/Brustwand. Heutzutage konzentrieren sich die Forscher entweder auf die Hypofraktionierung der adjuvanten Strahlentherapie (FAST-Forward-Studie [5] oder auf die Kombination von Chemotherapie und präoperativer Strahlentherapie. Die präoperative Strahlentherapie könnte zu einer Verbesserung des krankheitsfreien Überlebens und der Lebensqualität führen [6,7,8,9,10,11].
Die Hauptwirkung der Strahlung, insbesondere bei Strahlung mit niedrigem linearen Energietransfer, ist die Induktion reaktiver Sauerstoffspezies (ROS). Bei der Strahlentherapie entstehen ROS durch die Radiolyse von Wasser in der extrazellulären Umgebung, die für Tumorzellen und nahe gelegenes Normalgewebe toxisch sind. Etwa zwei Drittel der strahleninduzierten DNA-Schäden in Säugetierzellen werden auf ROS zurückgeführt [12]. Die Reaktion der Zellen auf strahleninduzierte DNA-Schäden ist stark von der Anwesenheit von Sauerstoff abhängig. Sauerstoffmoleküle können tatsächlich durch freie Radikale verursachte DNA-Schäden reparieren. Dies wird als „Sauerstoff-Fixierungshypothese“ bezeichnet [12,13]. In Abwesenheit von Sauerstoff werden DNA-Radikale durch Verbindungen mit Sulfhydrylgruppen reduziert, die die DNA in ihrer ursprünglichen Form reparieren. Nach dieser Hypothese ist die Hypoxie, d. h. der niedrige Sauerstoffgehalt im Tumor, eine der Hauptursachen für den klinischen Misserfolg der Strahlentherapie [14,15]. Hypoxie ist ein häufiges Merkmal der Mikroumgebung von Tumoren. ROS und Hypoxie sind zwei Faktoren mit entgegengesetzten Auswirkungen auf die Strahlenreaktion des Tumors [16]. Die allgemein akzeptierte Hypothese besagt, dass in hypoxischen Regionen des Tumors aufgrund des Mangels an ROS-Substrat Sauerstoff weniger oxidativer Stress auftritt. Jüngste Erkenntnisse haben jedoch gezeigt, dass die Zellen unter hypoxischen Bedingungen mehr ROS erzeugen, vor allem durch den mitochondrialen Stoffwechsel [17,18,19,20].
Ein charakteristisches Merkmal von Tumorzellen ist die Fähigkeit, ihren Stoffwechsel zu verändern und sie mit Energie und Stoffwechselprodukten zu versorgen, die sie für ihr Wachstum und Überleben unter nährstoff- und sauerstoffarmen Bedingungen benötigen. In Gegenwart vonO2 passen Krebszellen ihren Stoffwechsel jedoch auch an die Glykolyse an, indem sie die mitochondriale Pyruvat-Oxidation auf die Laktatproduktion umleiten [21,22]. Dieser Effekt wird als Warburg-Effekt bezeichnet. Jüngste Berichte deuten darauf hin, dass der Warburg-Effekt an der Resistenz gegen zytotoxischen Stress, der durch Chemo- oder Strahlentherapie ausgelöst wird, beteiligt ist [23,24,25,26,27]. Auf diese Weise können Behandlungsmethoden, die den glykolytischen Stoffwechsel blockieren oder reduzieren, die Empfindlichkeit der Tumorzellen gegenüber der Strahlentherapie erhöhen.
Unter hypoxischen Bedingungen bewirkt der Hypoxie-induzierbare Faktor 1-alpha (HIF1α) einen Anstieg der Expression von Pyruvat-Dehydrogenase-Kinasen (PDK1-4) [28]. Diese Enzyme sind für die Umschaltung des Stoffwechsels in den Mitochondrien verantwortlich, indem sie den Phosphorylierungsstatus (d. h. den Aktivitätszustand) der Pyruvatdehydrogenase (PDH) regulieren, die ein wichtiges Gatekeeper-Protein zwischen Glykolyse und mitochondrialer oxidativer Phosphorylierung (OXPHOS) ist. Dichloracetat (DCA), ein niedermolekularer PDK-Inhibitor, kann den Warburg-Effekt umkehren, indem er die PDH aktiviert und den Pyruvat-Stoffwechsel wieder in die Mitochondrien zurückverlagert. Die Hemmung der PDK durch DCA wird zur Behandlung von Laktatazidose und hereditären mitochondrialen Erkrankungen eingesetzt [29,30]. Insgesamt haben diese Beobachtungen dazu geführt, DCA als potenzielles Krebsmedikament zu betrachten [30,31].
DCA erhöht nachweislich die Strahlenempfindlichkeit von Dickdarm- und Prostatakrebszellen sowie von Plattenepithelkarzinomen der Speiseröhre und Glioblastomtumoren [32,33,34,35]. Bei Brustkrebszellen wurden jedoch keine ähnlichen Untersuchungen durchgeführt. Der Hauptmechanismus der Radiosensibilisierung in diesen Modellen wurde dem oxidativen Stress zugeschrieben. Gleichzeitig trugen auch der Zellzyklus-Stillstand in der G2-M-Phase und eine reduzierte mitochondriale Reservekapazität zu den radiosensibilisierenden Effekten bei. Auf der Grundlage der zuvor beschriebenen Ergebnisse werden derzeit zwei klinische Studien (eine bei Kopf-Hals-Karzinomen und eine bei Glioblastomen) durchgeführt, in denen die Antitumorwirkung von DCA in Kombination mit einer Strahlentherapie untersucht wird [36,37]. Alle präklinischen Untersuchungen wurden unter aeroben Bedingungen durchgeführt, aber es liegen keine Daten über die Auswirkungen einer DCA-Behandlung unter hypoxischen Bedingungen vor. In der vorliegenden Studie untersuchten wir daher zunächst die Hypothese, dass DCA das Laktat senkt und den Stoffwechsel von einem glykolytischen Phänotyp auf OXPHOS umstellt. Anschließend haben wir festgestellt, ob DCA hypoxische Brustkrebszellen radiosensibilisieren kann, und die zugrunde liegenden Mechanismen weiter untersucht. Die Ergebnisse dieser Studie könnten wichtige Auswirkungen auf klinische Studien haben, die darauf abzielen, PDK-Inhibitoren zur Verbesserung der Strahlenreaktion bei Brustkrebspatientinnen einzusetzen.
Ergebnisse
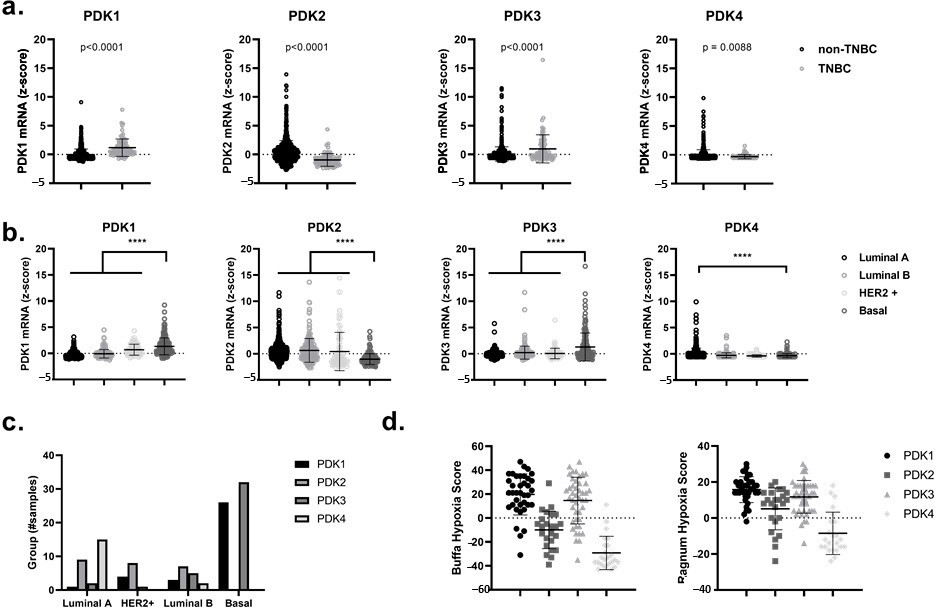
Hohe Expression von PDK1 und PDK3 bei Patienten mit dreifach negativem Brustkrebs (TNBC) und basalem Brustkrebs korreliert mit einer Hypoxie-bezogenen Gensignatur
Zunächst haben wir mit Hilfe des öffentlich zugänglichen cBioPortal for Cancer Genomics, analysierten wir zunächst die mRNA-Spiegel der vier verschiedenen PDK-Isomere, nämlich PDK1, PDK2, PDK3 und PDK4, in Patientendaten aus dem primären Brustkrebstumordatensatz TCGA (PanCancer Atlas and Cell 2015) [38,39]. Wir konnten eine signifikante Zunahme(p < 0,0001) der Expression von PDK1 und PDK3 bei TNBC im Vergleich zu Nicht-TNBC und bei basal-ähnlichen Brustkrebsen im Vergleich zu anderen Subtypen wie luminalen A-, luminalen B- und HER2-angereicherten Brustkrebsen nachweisen (Abbildung 1a-c). Bemerkenswert ist, dass die Hochregulierung von PDK1 und PDK3 bei Brustkrebspatientinnen mit höheren Ragnum- und Buffa-Hypoxie-Scores [40,41] korreliert sein könnte (Abbildung 1d). Diese Hypoxie-Scores beruhen auf der unterschiedlichen Expression spezifischer hypoxiebezogener Gene und sind über das cBioPortal for Cancer Genomics frei zugänglich. Die beobachtete Hochregulierung von PDK1 und PDK3 bei basal-ähnlichem Brustkrebs und ihre Korrelation mit einem hypoxischen Phänotyp (der mit der Aktivierung eines HIF1α-abhängigen Transkriptionsprogramms verbunden ist) legt nahe, dass der Einsatz von PDK-Inhibitoren eine attraktive therapeutische Modalität für die hypoxische Radiosensibilisierung darstellen könnte [42,43,44].
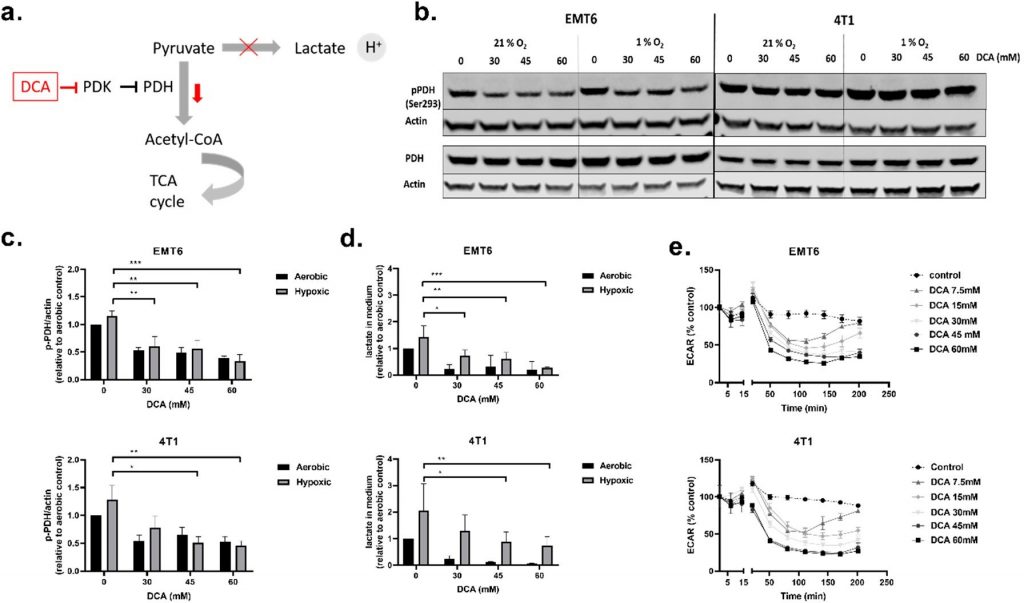
DCA verringerte die phosphorylierte PDH, den extrazellulären Laktatspiegel und die extrazelluläre Ansäuerungsrate (ECAR)
Wir begannen unsere In-vitro-Experimente mit der Durchführung von Lebensfähigkeitstests (Abbildung S1a, b), um die wachstumshemmenden Eigenschaften von DCA zu bestimmen. DCA verringerte die Lebensfähigkeit der Zellen in EMT6- und 4T1-Krebszellen dosisabhängig und unabhängig vomO2-Status (Abbildung S1a). Ein Proliferationsassay zeigte außerdem, dass mit steigender DCA-Konzentration eine Verschiebung von der Wachstumsverzögerung hin zu zytostatischen und sogar zytotoxischen Wirkungen zu beobachten war (Abbildung S1b).
Als nächstes untersuchten wir den Einfluss von DCA auf die PDK1-4-Aktivität und den Stoffwechsel von TNBC-Zellen. PDH ist ein wichtiges Gatekeeper-Protein der Glykolyse und der mitochondrialen OXPHOS, d. h. PDH katalysiert die ratenbegrenzende Decarboxylierung von Pyruvat zu Acetyl-CoA. PDH wird durch Phosphorylierung (an Ser293) durch PDK gehemmt, und diese Hemmung kann durch Dephosphorylierung durch Pyruvatdehydrogenase-Phosphatase (PDP) aufgehoben werden [45,46] (Abbildung 2a). Bei akzeptablen Toxizitätsdosen (30 mM, 45 mM und 60 mM) bewerteten wir die Wirkung von DCA auf die PDK-Aktivität, indem wir den Gehalt an phosphorylierter PDH (p-PDH), Laktat im extrazellulären Medium und die ECAR der Zellen in Echtzeit maßen (Abbildung 2b-e). Alle drei DCA-Dosen verringerten die Menge an p-PDH in EMT6- und 4T1-Zellen sowohl unter sauerstoffhaltigen als auch unter hypoxischen Bedingungen in dosisabhängiger Weise. Der Rückgang von p-PDH war bei EMT6 für alle DCA-Dosen signifikant, während bei 4T1 nur 45 mM und 60 mM p-PDH unter hypoxischen Bedingungen signifikant verringerten (Abbildung 2b,c). Bei der Bewertung der Wirkung niedrigerer DCA-Dosen stellten wir fest, dass die Menge an p-PDH in EMT6-Zellen zu sinken beginnt, wenn sie mit 3 mM DCA behandelt werden, und in 4T1-Zellen, wenn sie mit 10 mM DCA behandelt werden (Abbildung S2a,b). Die Menge an Laktat im Medium war unter hypoxischen Bedingungen im Vergleich zu aeroben Bedingungen in beiden Zelllinien erhöht, was für einen Nettoanstieg des glykolytischen Umsatzes in Zellenmit O2-Entzug spricht [47,48]. In Übereinstimmung mit den Western-Blot-Ergebnissen führte die Behandlung mit DCA bei beiden Zelllinien zu einem dosisabhängigen Rückgang des Laktats im Medium (Abbildung 2d). Obwohl die Verringerung der Laktatfreisetzung unter aeroben Bedingungen dramatisch war, wurde eine dosisabhängige Verringerung der Produktion des glykolytischen Endprodukts auch unter Hypoxie beobachtet. Schließlich verursachte DCA ab einer Dosis von 7,5 mM eine zeitabhängige Verringerung der ECAR sowohl in EMT6 als auch in 4T1 (Abbildung 2e). Der anfängliche Rückgang der ECAR nach der Behandlung mit Dosen unter 30 mM DCA wurde bei EMT6 nach 2,5 Stunden ausgeglichen. Bei 4T1 beobachteten wir denselben Effekt bei DCA-Dosen unter 15 mM. Diese Ergebnisse zeigen, dass die Behandlung von TNBC-Zelllinien der Maus mit DCA die PDH-Phosphorylierung hemmt und das Ausmaß der Glykolyse sowohl unter aeroben als auch unter hypoxischen Bedingungen verringert.
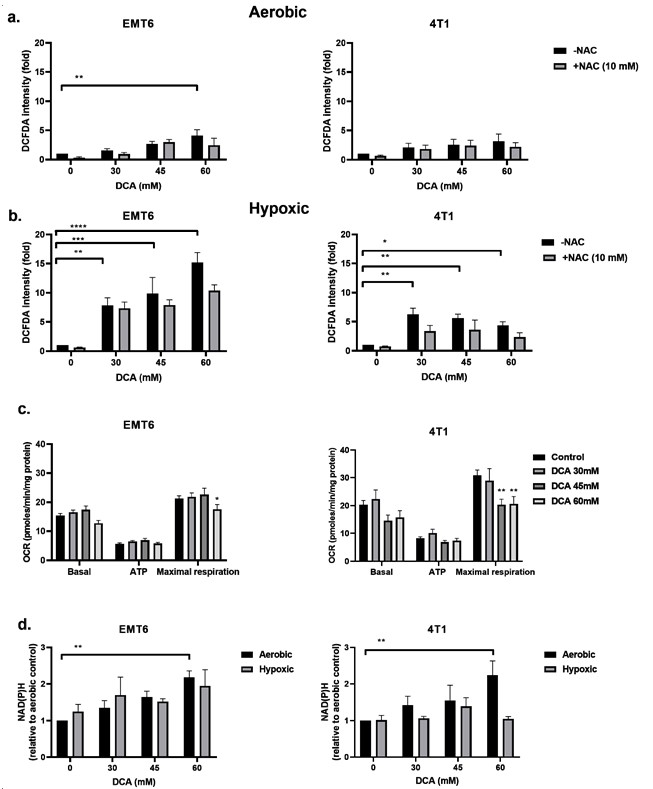
DCA induziert die ROS-Produktion in hypoxischen Krebszellen
Es wird berichtet, dass die Hemmung der PDK und die Aktivierung der PDH mit einer Hochregulierung der intrazellulären ROS einhergeht [49,50]. ROS ist von zentraler Bedeutung für die Entstehung von DNA-Schäden nach einer Bestrahlung. Wir untersuchten die ROS-Konzentrationen in EMT6- und 4T1-Zellen unter aeroben und hypoxischen Bedingungen mit Hilfe der CM-H2DCFDA-Sonde. Wie in Abbildung 3a,b dargestellt, löste DCA sowohl in EMT6 als auch in 4T1 eine dosisabhängige ROS-Produktion aus. Unter aeroben Bedingungen führte nur die höchste DCA-Dosis (60 mM) zu einem signifikanten ROS-Anstieg um das bis zu Fünffache im Vergleich zur Kontrolle bei EMT6-Zellen. Bei 4T1-Zellen wurde unter aeroben Bedingungen kein signifikanter Anstieg von ROS festgestellt. Die Erhöhung der ROS-Konzentration wurde durch die Zugabe des ROS-Fängers N-Acetyl-Cystein (NAC) zum Teil wieder aufgehoben. Unter hypoxischen Bedingungen wiesen wir einen dosisabhängigen Anstieg von ROS nach, der bei EMT6-Zellen das 15-fache und bei 4T1-Zellen das 5-fache erreichte; die niedrigste DCA-Dosis führte bei beiden Zelltypen sogar zu einem signifikanten Anstieg von ROS (Abbildung 3b).
Wir schlussfolgerten, dass die erhöhte ROS-Produktion unter Hypoxie aus den kombinierten Effekten der veränderten mitochondrialen Elektronentransportkette (aufgrund der Verringerung vonO2 als letztem Elektronenakzeptor) und dem erzwungenen DCA-getriebenen oxidativen Pyruvat-Stoffwechsel resultieren könnte. Mit Hilfe des Seahorse-Analysegeräts stellten wir fest, dass DCA keinen Einfluss auf die Basalatmung und die ATP-Produktion hatte, aber die maximale Atmungskapazität in EMT6- und 4T1-Tumorzellen deutlich verringerte (Abbildung 3c). Eine andere Möglichkeit ist, dass DCA den NAD(P)H-Spiegel erhöht, was mit einer erhöhten ROS-Produktion verbunden ist. Wir sahen, dass die Behandlung mit DCA unter aeroben Bedingungen einen dosisabhängigen Anstieg von NAD(P)H in EMT6- und 4T1-Zellen zur Folge hatte (Abbildung 3d). Unter hypoxischen Bedingungen zeigten wir einen möglichen Anstieg von NAD(P)H in EMT6-Zellen, die mit 60 mM DCA behandelt wurden, aber keinen Anstieg in 4T1-Zellen (Abbildung 3d). Zusammen mit den oben genannten ROS-Daten deuten diese Ergebnisse darauf hin, dass der mitochondriale Stoffwechsel zwar erhalten bleibt, aber ein lokaler Anstieg der ROS-Produktion bei DCA-Exposition die Integrität und Funktion der Mitochondrien verändern kann.
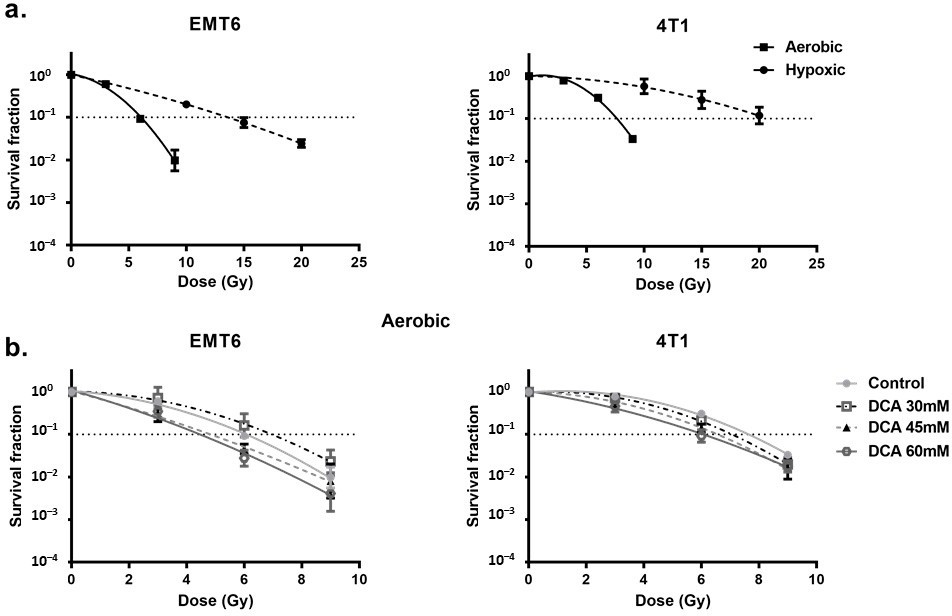
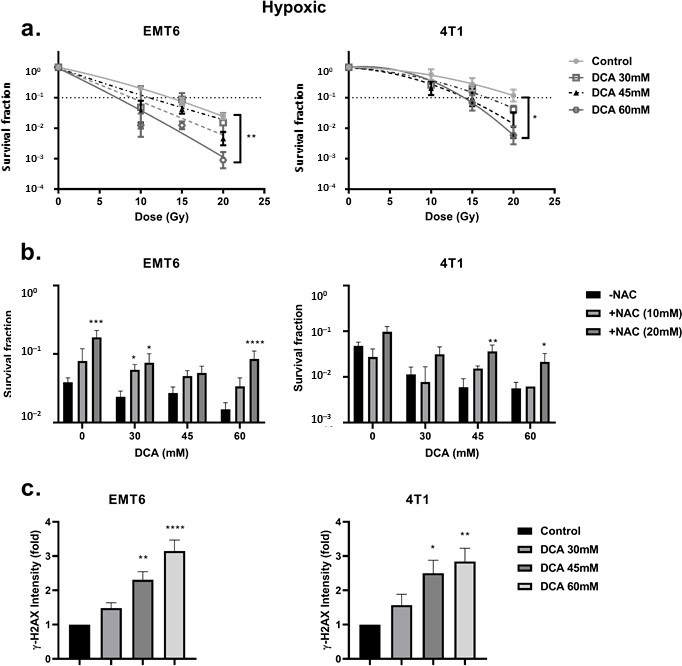
DCA radiosensibilisiert hypoxische Zellen durch ROS
Zunächst untersuchten wir die Hypoxie-induzierte Radioresistenz der Zellen. Beim Vergleich von hypoxischen mit aeroben Bedingungen stellten wir eine stark beeinträchtigte Radioresistenz fest, mit einem Sauerstoffanreicherungsverhältnis von 2,7 bzw. 2,3 für EMT6- und 4T1-Tumorzellen (Abbildung 4a). In diesem Zusammenhang beobachteten wir, dass die DCA-Behandlung einen kleinen Effekt der intrinsischen Radiosensibilisierung in EMT6-, nicht aber in 4T1-Zellen bewirkte (Abbildung 4b). Interessanterweise überwand 60 mM DCA in Übereinstimmung mit den Ergebnissen der ROS-Erzeugung unter hypoxischen Bedingungen die hypoxische Radioresistenz signifikant(p < 0,05) mit Verstärkungsverhältnissen von 2,3 und 1,5 bei 60 mM für EMT6- bzw. 4T1-Tumorzellen (Abbildung 5a). Die radiosensibilisierende Wirkung wurde durch NAC sowohl bei EMT6- als auch bei 4T1-Zellen aufgehoben (Abbildung 5b). Die Hauptursache für den strahleninduzierten Zelltod durch ROS liegt in der Induktion von Doppelstrangbrüchen in der DNA (ds-DNA) [12,51]. Wir untersuchten daher die ds-DNA-Schäden nach der Behandlung mit DCA durch Quantifizierung des Phosphorylierungsstatus von γH2AX unter hypoxischen Bedingungen. Wie in Abbildung 5c dargestellt, erhöhte DCA die Bildung von ds-DNA-Schäden sowohl in EMT6 als auch in 4T1 in einer dosisabhängigen Weise.
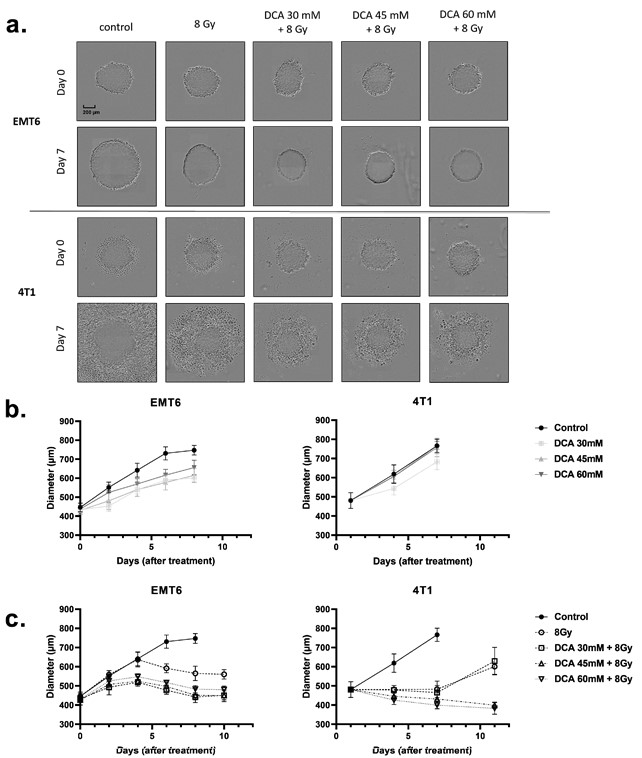
DCA radiosensibilisiert 3D-Zellkulturen (Sphäroide)
Die obigen Ergebnisse veranlassten uns zu untersuchen, ob DCA auch die Radiosensitivität von dreidimensionalen (3D) Zellkulturmodellen (Abbildung 6a-c) verbessern könnte, die die physikalisch-chemischen Eigenschaften der Mikroumgebung des Tumors, einschließlich Sauerstoffgradienten, besser nachahmen. Anhand von Sphäroiden, die aus EMT6- und 4T1-Zellkulturen gewonnen wurden, haben wir das Sphäroidwachstum nach DCA-Behandlung und Strahlentherapie gemessen (Abbildung 6a). Die DCA-Behandlung allein führte zu einer geringen Wachstumsverzögerung bei den EMT6-Zellen, veränderte jedoch nicht das Wachstum der 4T1-Sphäroide (Abbildung 6b). Eine Bestrahlung mit 8 Gy reduzierte das Wachstum der Tumor-Sphäroide, ein Effekt, der durch die Kombination mit DCA-Behandlung noch verstärkt wurde (Abbildung 6a,c). Bemerkenswert ist, dass in 4T1-Sphäroiden zytotoxische Wirkungen beobachtet wurden (erkennbar an den Halos toter Zellen), während in EMT6-Sphäroiden zytostatische Wirkungen beobachtet wurden (Abbildung 6a).
Die Kombination von DCA und Strahlentherapie verzögert das Tumorwachstum in vivo nicht
Als Nächstes untersuchten wir, ob der in vitro-Nutzen der DCA-Kombination mit der Strahlentherapie auch in vivo bestätigt werden kann (Abbildung S3a-d). Mäuse, denen EMT6- oder 4T1-Brustkrebszellen injiziert worden waren, wurden entweder einer einmaligen (12 Gy bzw. 15 Gy) (Abbildung S3a,c) oder fraktionierten (5*4 Gy bzw. 5*6 Gy) (Abbildung S3b,d) Bestrahlung ausgesetzt. Die einzelnen und fraktionierten Bestrahlungsdosen pro Tumortyp sind in Bezug auf die biologisch effektive Dosis (BED) ähnlich und unterscheiden sich in Abhängigkeit von der intrinsischen Radiosensitivität der verwendeten Zelllinien. Wichtig ist, dass die Injektion von DCA entweder i.p. oder i.t. für 10 Tage sicher war, ohne eine merkliche Toxizität zu verursachen (Abbildung S4a-d). Die Bestrahlung allein verzögerte das Tumorwachstum bei EMT6 sieben Tage lang mit einer einzigen Fraktion und vier Tage bei fraktionierter Bestrahlung (Abbildung S3a,b). Bei 4T1-Tumoren verzögerte die Bestrahlung das Tumorwachstum erwartungsgemäß um fünf Tage bei einer einzelnen Fraktion und um 10 Tage bei einer fraktionierten Bestrahlung (Abbildung S3c,d). DCA (300 mg/kg), entweder durch intraperitoneale (ip) oder intratumorale (it) Injektion, verzögerte das Tumorwachstum nicht, ebenso wenig wie die Kombination von DCA mit Strahlung (Abbildung S3a-d). Als Nächstes überprüften wir, ob die Behandlung mit DCA eine Hypoxie in den Tumoren auslösen könnte (Abbildung S3e,f). Obwohl das Ausmaß der mit Pimonidazol gefärbten Hypoxie in EMT6-Tumoren nicht verändert war, wurde als Reaktion auf DCA in 4T1-Tumoren ein Trend zu einer Abnahme der Hypoxie beobachtet.
Diskussion
In dieser Studie sollte untersucht werden, ob DCA, das in den mitochondrialen Stoffwechsel eingreift, TNBC- und basalähnliche Brustkrebszellen für eine Strahlentherapie sensibilisieren könnte. Bei den meisten TNBC- und basalähnlichen Brustkrebsarten handelt es sich um aggressive Tumore, für die es nur begrenzte Behandlungsmöglichkeiten gibt und die eine schlechte Prognose haben [2,3,4]. Diese Tumore weisen einen verstärkten glykolytischen Phänotyp auf, der ihre schlechte Prognose unterstützt und zusätzlich mit der Strahlenresistenz korreliert ist [52]. In der aktuellen Studie fanden wir heraus, dass die mRNA-Konzentrationen für zwei der vier PDK-Isoformen (PDK1 und PDK3) bei TNBC und basalähnlichen Brustkrebs-Subtypen hochreguliert sind. Die Überexpression von PDKs wurde in zahlreichen menschlichen Tumorproben nachgewiesen [42,53,54,55,56,57,58,59,60,61], und viele Krebszelllinien weisen eine erhebliche Hochregulierung von PDK-Isoformen auf [50,62,63]. Es wurde berichtet, dass eine Überexpression von PDK bei einer Vielzahl von Tumorarten mit einer schlechten Prognose verbunden ist [53,54,55,56,57,58,59,60,61]. Die Überexpression von PDKs in Krebszellen wird von verschiedenen Transkriptionsfaktoren, wie z. B. HIF1, beeinflusst [28,64]. HIF1 unterdrückt aktiv die OXPHOS, indem es die Gene, die für PDK1 und PDK3 kodieren, direkt transaktiviert. PDK wiederum phosphorylieren und inaktivieren PDH [42]. Die Hochregulierung der PDKs bei Krebs kann also unmittelbar auf transformierende Mutationen und die hypoxische Mikroumgebung des Tumors zurückgeführt werden. In Übereinstimmung mit diesen Erkenntnissen beobachteten wir, dass die Hochregulierung von PDK1 und PDK3 mRNA mit hypoxiebezogenen Genprofilen korreliert. Eine metabolische Umprogrammierung durch gezielte Ansteuerung der PDK-Enzyme, um von der Glykolyse auf die OXPHOS umzuschalten, scheint daher ein vielversprechender therapeutischer Weg zur Behandlung von Brustkrebs mit begrenzten Therapiemöglichkeiten zu sein.
Das wichtigste Ergebnis unserer Studie ist, dass der PDK-Inhibitor DCA die glykolytische Aktivität von Brustkrebszellen in Gegenwart von Sauerstoff, aber auch unter Hypoxie verringern kann [29]. Wir fanden heraus, dass DCA die Menge der phosphorylierten PDH senkt und einen dosisabhängigen Rückgang der extrazellulären Laktatwerte und der ECAR in aeroben und hypoxischen Zellen auslöst. Als nächstes kombinierten wir DCA und Strahlentherapie unter der Hypothese, dass Tumorzellen durch die Umkehrung des glykolytischen Phänotyps und die Umleitung von mehr Pyruvat in die mitochondriale Oxidation mehr ROS produzieren und empfindlicher gegenüber Strahlung werden könnten. Intrinsische radiosensibilisierende Wirkungen von DCA wurden bereits für Glioblastomzellen [34,35]zellen des nicht-kleinzelligen Lungenkarzinoms (NSCLC) [65,66]kolorektale [35]prostatakrebszellen [32] und radioresistente Medulloblastomzellen [67]. Die vorgeschlagenen Mechanismen sind Zellzyklus-Stillstand in der G2-M-Phase, wodurch zusätzliche DNA-Schäden und ein konsekutiver Zelltod als Reaktion auf die erhöhte mitochondriale ROS-Produktion entstehen. In der aktuellen Studie haben wir festgestellt, dass DCA unter aeroben Bedingungen nur minimale radiosensibilisierende Wirkungen bei der höchsten ungiftigen DCA-Konzentration zeigte, während DCA hypoxische Brustkrebszellen sowohl in 2D- als auch in 3D-Sphäroiden stark radiosensibilisierte. Obwohl ROS theoretisch mit oxidativen Mechanismen verbunden sind, besteht eine Partnerschaft zwischen Hypoxie und ROS in Tumoren. Hypoxie erhöht die ROS-Bildung durch Verlängerung der Lebensdauer der Semichinon-Radikale; umgekehrt helfen ROS den Tumorzellen bei der Anpassung an Hypoxie durch Stabilisierung von HIF1-α [16,68]. Extrazelluläre Insulte könnten diese Partnerschaft jedoch unterbrechen, indem sie eine übermäßige ROS-Produktion auslösen, die die mitochondriale Atmung beeinträchtigt und so den hypoxischen Anteil in Tumoren verringert [16,69,70]. In diesem Zusammenhang hemmt Arsentrioxid den Sauerstoffverbrauch von Tumorzellen durch einen Anstieg der intrazellulären ROS, was zu einer verstärkten Radioresponse führt [71]. Auch die Unterdrückung der Glykolyse soll die Strahlenreaktion erhöhen. Dies kann durch Ritonavir (Glukosetransporter-Inhibitor), 2-Desoxyglukose (Hexokinase-Inhibitor) und Lonidamin (Hexokinase-Inhibitor) geschehen, die derzeit in klinischen Studien bei verschiedenen Krebsarten untersucht werden [72,73,74,75]. Eine weitere Möglichkeit besteht darin, dass nach der Behandlung mit DCA aufgrund der Induktion der NADPH-Oxidase ROS gebildet werden [76]. Es wurden jedoch keine direkten Beweise gefunden, die darauf schließen lassen, dass DCA die NADPH-Oxidase hochregulieren kann [77]. Wir beobachteten einen dosisabhängigen Anstieg von NAD(P)H unter aeroben Bedingungen, aber nicht unter hypoxischen Bedingungen in EMT6- und 4T1-Zellen. Wir stellen daher die Hypothese auf, dass der Anstieg von NAD(P)H und die Hochregulierung von NADPH-Oxidasen nur eine untergeordnete Rolle bei der Zunahme von ROS unter hypoxischen Bedingungen spielt. Unserer Ansicht nach ist der primäre Mechanismus der beobachteten radiosensibilisierenden Effekte sehr wahrscheinlich auf die mehrfache Steigerung der ROS-Produktion (bis zum 15-fachen) nach DCA-Behandlung unter hypoxischen Bedingungen zurückzuführen.
In Übereinstimmung mit der Literatur haben wir gezeigt, dass supraphysiologische Konzentrationen von DCA erforderlich sind, um Veränderungen der Stoffwechselaktivität, eine erhöhte ROS-Bildung und eine Radiosensibilisierung hervorzurufen [29]. Unsere zentrale Hypothese ist, dass diese Effekte die Folge der PDK-Hemmung durch DCA sind [78]. Die Konzentrationen, die erforderlich sind, um die gemessenen Ergebnisse hervorzurufen, sind jedoch um ein Vielfaches höher als die Hemmkonstante (Ki) von PDK1-4. Es ist bemerkenswert, dass DCA physiologischerweise als Anion existiert, trotz seiner geringen Größe relativ membranundurchlässig ist und daher den mitochondrialen Pyruvat-Träger für die mitochondriale Aufnahme benötigt [79,80]. Die Konjugation von DCA an einen lipophilen Träger verbesserte jedoch den mitochondrialen Transport. Dadurch wurde der IC50-Wert von DCA von einem millimolaren auf einen niedrigen mikromolaren Bereich gesenkt, was deutlich innerhalb des Ki-Bereichs von PDK1-4 liegt [81]. DCA ahmt die wirksame Hemmung von PDK1-4 durch siRNA nach, und DCA, das zu PDK-siRNA hinzugefügt wurde, hatte keine zusätzlichen Auswirkungen [49,50,60,82,83,84,85,86,87,88]. Darüber hinaus könnte ein DCA-ähnliches kleines Molekül direkt oder indirekt andere zelluläre und molekulare Ziele beeinflussen. Kürzlich durchgeführte Untersuchungen ergaben, dass eine DCA-Behandlung die Konzentration aller TCA-Zwischenprodukte erhöhte, aber weder die Glukoseaufnahme noch die Glykolyse beeinflusste [89]. Andere Forscher zeigten Hinweise darauf, dass DCA die de novo CoA-Biosynthese steigern kann. Da hohe CoA-Konzentrationen für Zellen toxisch sein können, könnte dieser metabolische Effekt teilweise für die durch DCA vermittelte Toxizität für Krebszellen verantwortlich sein [90]. Jüngste Forschungsarbeiten haben eine neue Hypothese aufgestellt, die besagt, dass die Wirksamkeit von DCA gegen Krebs möglicherweise auf seiner Fähigkeit beruht, Acetat zu bekämpfen. Hohe Acetatspiegel können die DNA-, RNA- und Proteinsynthese fördern. Außerdem kann es mit einer Resistenz gegen Krebsmedikamente in Verbindung gebracht werden [91]. Schließlich fanden Forscher heraus, dass DCA den AMPK-Signalweg aktivieren kann, was zu einer Kaskade von nachgelagerten metabolischen und krebsbekämpfenden Wirkungen führt [92,93]. Dennoch bleibt unsere Hypothese bestehen, dass unter hypoxischen Bedingungen die Zufuhr von Pyruvat in die Mitochondrien eine Hochregulierung der ROS-Konzentration bewirkt, die Krebszellen radiosensibilisiert. Es ist uns nicht gelungen, diese Effekte in vivo zu rekapitulieren, und es sind weitere Arbeiten erforderlich, um festzustellen, wie die radiosensibilisierenden Effekte von DCA in hypoxischen Tumorkompartimenten umgesetzt werden können. Pharmakokinetische Probleme im Zusammenhang mit der DCA-Verabreichung in vivo können ausgeschlossen werden, und die verwendete DCA-Dosis (150 mg/kg) liegt deutlich unter den in der Literatur verwendeten Dosen [29]. Die menschliche Äquivalentdosis der von uns in vivo verwendeten DCA-Dosis beträgt 12 mg/kg/d und liegt damit deutlich innerhalb des in klinischen Studien verwendeten Toleranzbereichs. Eine mögliche Erklärung für unsere fehlgeschlagenen In-vivo-Experimente ist, dass eine höhere DCA-Dosis erforderlich ist, um in vivo eine radiostimulierende Wirkung zu erzielen. In den letzten 30 Jahren wurde DCA in der Tat mit angemessenem Erfolg als Prüfpräparat zur Behandlung von Typ-2-Diabetes, erworbener und angeborener Hyperlipoproteinämie, Myokardischämie, erworbener und angeborener Laktatazidose und in jüngster Zeit auch von Krebs verabreicht [29,30]. In mehreren Phase-I/II-Studien werden die Sicherheit von DCA und seine Wirkung als Krebsmittel untersucht. DCA wird schnell absorbiert und kann sogar die Blut-Hirn-Schranke überwinden. In zwei Phase-I-Studien wurde die Sicherheit von oral verabreichtem DCA bei Patienten mit rezidivierenden bösartigen Hirntumoren oder Hirnmetastasen von Krebserkrankungen, die nicht das zentrale Nervensystem betreffen, untersucht [94,95]. Diese Studien zeigten, dass DCA im Allgemeinen von den Patienten gut vertragen wird. Eine andere Erklärung für den rätselhaften Unterschied zwischen In-vitro- und In-vivo-Effekten ist eher auf Veränderungen des metabolischen Phänotyps zurückzuführen, wenn Krebszellen (ektopisch) in vivo injiziert werden. In der Tat wird die erhöhte In-vitro-Radiosensitivität durch DCA unter Bedingungen von Hypoxie und hohem glykolytischem Stoffwechsel erreicht, so dass die Verschiebung von Glykolyse zu Oxphos bei DCA-Behandlung zusammen mit einer zusätzlichen ROS-Hochregulierung beobachtet werden kann.
Das begrenzte Ausmaß der Hypoxie in In-vivo-Tumoren könnte daher auf eine eingeschränkte Fähigkeit von DCA zurückzuführen sein, eine Veränderung zu bewirken, die bereits in gut mit Sauerstoff versorgten Tumoren vorhanden ist, die weitgehend von der OXPHOS abhängen. Weitere Untersuchungen sind erforderlich, um die radiosensibilisierende Wirkung von DCA in Brusttumormodellen der Maus zu testen, die durch eine begrenzte Angiogenese und erhöhte hypoxische Anteile gekennzeichnet sind.
Zusammenfassend zeigen wir, dass DCA die hypoxische Radioresistenz von Brustkrebszellen in 2D- und 3D-Systemen überwindet, was in erster Linie auf die Hochregulierung von ROS zurückgeführt werden kann. Bemerkenswerterweise führt die DCA-induzierte Verschiebung des Glykolyse-zu-OXPHOS-Stoffwechsels auch zu oxidativem Stress unter hypoxischen Bedingungen. DCA wird seit vielen Jahren zur Behandlung von Stoffwechselkrankheiten und vererbten mitochondrialen Erkrankungen eingesetzt. In den letzten zehn Jahren wurde DCA weitgehend als Krebsmedikament umgewidmet, mit vielversprechenden präklinischen Daten, Fallberichten und klinischen Studien, wie zuvor beschrieben. Die aktuellen präklinischen Ergebnisse deuten darauf hin, dass eine weitere Untersuchung des krebsbekämpfenden Potenzials von DCA notwendig ist, wenn man bedenkt, dass hypoxische Tumorzellen von dem PDK-Inhibitor nicht verschont bleiben, der tödlichen oxidativen Stress auslösen kann, insbesondere in Kombination mit einer Strahlentherapie.
Materialien und Methoden
TCGA Breast Cancer Cohort Analysis
Die PDK1-4 mRNA-Expressionsprofile (RNA Seq V2 RSEM oder log RNA Seq V2 RSEM) wurden von der cBioPortal-Website in Form von z-score-transformierten Daten abgefragt [38,39]. Die abgefragten Daten wurden aus 1084 öffentlich verfügbaren Brustkrebsfällen des TCGA PanCancer Atlas und aus 817 Brustkrebsfällen der TCGA Cell 2015 Datenbank ausgewertet. Für den TCGA Cell 2015-Datensatz wurde die Analyse von PDK1-4 durchgeführt, indem die dreifach negative Subpopulation mit dem Rest der Brustkrebsfälle verglichen wurde. Triple-negativer Brustkrebs wurde durch einen „negativen“ Status für die immunhistochemischen Scores der Gene Östrogenrezeptor (ER), Progesteronrezeptor (PR) und humaner epidermaler Wachstumsfaktor-Rezeptor 2 (HER2) bestimmt (insgesamt 83 Fälle). In der TCGA PanCancer Atlas-Datenbank wurde die Analyse der PDK1-4-Expression in verschiedenen Pam50-Kategorien durchgeführt. Pam50 ist eine 50-Gene-Signatur, die Brustkrebs in fünf molekulare intrinsische Subtypen einteilt: Luminal A, Luminal B, HER2-angereichert, Basal-like und Normal-like. Die mRNA-Expression von TCGA-Brustkrebs und die klinischen Daten wurden direkt auf der cBioPortal-Website analysiert oder für weitere Analysen heruntergeladen. Die Buffa- und Ragnum-Hypoxie-Score-Analyse der Proben, bei denen die PDK1-4-Expression einen z-Score von mehr als 2 aufweist, wurde direkt auf der cBioPortal-Website durchgeführt.
Zelllinien und Chemikalien
Die EMT6-Zelllinie des Mamma-Adenokarzinoms wurde freundlicherweise von Edith Lord (University of Rochester, Cancer Center, New York) zur Verfügung gestellt, und 4T1-Zellen wurden von der American Type Culture Collection bezogen. Alle Experimente wurden in Roswell Park Memorial Institute 1640 Medium (Thermo Fisher Scientific, Waltham, MA, USA) durchgeführt, das mit 10% fötalem Rinderserum (Greiner Bio-One, Kremsmünster, Österreich) ergänzt wurde. Für alle Behandlungen der Zellen wurde HEPES-Puffer verwendet. Die Chemikalien wurden, sofern nicht anders angegeben, von Sigma-Aldrich (Sigma-Aldrich, St. Louis, MO, USA) bezogen
Behandlungen
EMT6 und 4T1 wurden bis zur Konfluenz gezüchtet und 16 Stunden lang mit DCA in den angegebenen Konzentrationen behandelt. N-Acetylcystein (NAC) wurde in einer Konzentration von 10 mM oder 20 mM sowohl 1 Stunde vor als auch während der Behandlung mit DCA zu den Kulturen gegeben. Danach wurden die Kulturen wie unten beschrieben für weitere Analysen verwendet. Die Behandlung erfolgte entweder unter aeroben Bedingungen oder unter hypoxischen Bedingungen. Hypoxie wurde durch Inkubation in einem Stickstoff/Kohlendioxid-Gleichgewicht mit 1 % Sauerstoff induziert [96]
MTT-Assay
Die Zytotoxizität von DCA wurde mittels MTT-Assay bewertet, wie an anderer Stelle <a href=“#97″>[97,</a></sup><a href=“#98″><sup>98]</sup> beschrieben .</a> Kurz gesagt wurden die Zellen in 96-Well-Platten gezüchtet und mit den angegebenen Konzentrationen behandelt. Nach der Behandlung wurde das Medium abgesaugt und 50 µL des MTT-Reagens (5 mg/mL) für 1,5 h zugegeben. Anschließend wurden 200 µL MTT-Lösungsmittel (19:1 DMSO/HCL) zugegeben und gemischt, um die in den Zellen gebildeten Formazankristalle aufzulösen. Die Absorption wurde bei einer Wellenlänge von 540 nm mit einem Spektrophotometer (Bio-Rad Laboratories, Hercules, CA, USA) gemessen. Die Lebensfähigkeit der Zellen wurde durch Normalisierung der behandelten Zellen auf die unbehandelten Kontrollzellen bestimmt.</p>