Allison B. Haugrud, Yongxian Zhuang, Joseph D. Coppock, W. Keith Miskimins
Forschungszentrum für Krebsbiologie, Sanford Research, 2301 E. 60th St North, Sioux Falls, SD 57104, USAe-mail
: [email protected]
Received: 24 April
2014Akzeptiert: 5. September 2014Veröffentlicht
: 12. September 2014
Zusammenfassung
Der einzigartige Stoffwechsel von Brustkrebszellen macht es interessant, dieses Phänomen therapeutisch zu nutzen. Metformin, ein vielversprechendes Brustkrebstherapeutikum, greift den Komplex I der Elektronentransportkette an, was zu einer Anhäufung reaktiver Sauerstoffspezies (ROS) führt, die schließlich zum Zelltod führen. Die Hemmung von Komplex I führt zur Produktion von Laktat, einem metabolischen Nebenprodukt, das bereits in hohem Maße von umprogrammierten Krebszellen produziert wird und mit einer schlechten Prognose verbunden ist. Während Metformin ein vielversprechendes Krebstherapeutikum bleibt, haben wir nach einem ergänzenden Wirkstoff gesucht, der die apoptosefördernde Wirkung von Metformin verstärkt und gleichzeitig die Laktatproduktion abschwächt, was zu einer deutlich verbesserten Wirksamkeit führen könnte. Dichloracetat (DCA) ist ein bewährtes Medikament zur Behandlung von Laktatazidose, das durch Hemmung der Pyruvat-Dehydrogenase-Kinase (PDK) wirkt und den mitochondrialen Stoffwechsel fördert. Unser Ziel war es, die Synergie und die Mechanismen zu untersuchen, durch die diese beiden Medikamente Brustkrebszellen abtöten. Die Zelllinien wurden den angegebenen Behandlungen unterzogen und auf Zelltod und verschiedene Aspekte des Stoffwechsels untersucht. Der Zelltod und die ROS-Produktion wurden mittels Durchflusszytometrie, Western-Blot-Analyse und Zellzählung analysiert. Bilder der Zellen wurden mit Phasenkontrastmikroskopie oder konfokaler Mikroskopie aufgenommen. Der Stoffwechsel der Zellen wurde mit dem Seahorse XF24-Analysegerät, Laktat-Assays und pH-Analysen untersucht. Wir zeigen, dass die Kombination von DCA und Metformin zu einer synergistischen Induktion der Apoptose von Brustkrebszellen führt. Metformin-induzierte oxidative Schäden werden durch DCA durch PDK1-Hemmung verstärkt, wodurch auch die durch Metformin geförderte Laktatproduktion verringert wird. Wir zeigen, dass DCA und Metformin in Kombination auf synergistische Weise eine Caspase-abhängige Apoptose auslösen, die mit oxidativen Schäden einhergeht, und gleichzeitig die von Metformin geförderte Laktatproduktion abschwächen. Innovative Kombinationen wie Metformin und DCA sind vielversprechend für die Erweiterung der Brustkrebstherapien.
Schlüsselwörter: Metformin; Dichloroacetat; Brustkrebs; Laktat; Apoptose
© Springer Science+Business Media New York 2014
EINFÜHRUNG
Der Krebsstoffwechsel entwickelt sich zu einem vielversprechenden Gebiet für die Entwicklung neuer therapeutischer Ansätze. Im Vergleich zu den normalen Zellen, aus denen sie hervorgegangen sind, sind Krebszellen metabolisch umprogrammiert und nutzen die Glykolyse auch bei ausreichendem Sauerstoffangebot bevorzugt, ein Phänomen, das als Warburg-Effekt bekannt ist [1]. Um den ATP-Verlust infolge der bevorzugten Glykolyse (im Gegensatz zur oxidativen Phosphorylierung) zu kompensieren, regulieren Krebszellen Gene, die für Glukosetransporter und glykolytische Enzyme wie die Pyruvat-Dehydrogenase-Kinase (PDK) und die Laktat-Dehydrogenase (LDH) kodieren, hoch. Diese hohe Rate der Glukoseaufnahme und der veränderte Stoffwechsel liefern nicht nur ATP und ermöglichen den Zellen das Überleben unter hypoxischen Bedingungen, sondern liefern auch biosynthetische Bausteine wie Zwischenprodukte und Substrate für die Produktion von Aminosäuren, NADPH und Ribose-5-Phosphat, die für die Nukleotid-, Protein- und Membransynthese in sich schnell teilenden Zellen unerlässlich sind. Dies bedeutet auch, dass der mitochondriale TCA-Zyklus einen geringeren Prozentsatz an ATP erzeugt, so dass Citrat für die Fettsäure- und Lipidbiosynthese zur Herstellung neuer Membranen verwendet werden kann [2]. Ein großer Teil des aus der Glykolyse stammenden Laktats wird von den umliegenden Zellen aufgenommen, die sich gegenseitig recyceln, um das Wachstum des Tumors und die Resistenz gegen apoptotische Zelltodmechanismen zu unterstützen [2, 3]. Das von Tumoren produzierte Laktat kann den T-Zell-Stoffwechsel und die Antigenpräsentation dendritischer Zellen stören, was zu einer Umgehung der Immunabwehr des Tumors führt [4, 5]. Der hohe Glukoseverbrauch und die Glykolyse verschaffen den Krebszellen also zahlreiche Vorteile. Es könnte jedoch auch möglich sein, das einzigartige Stoffwechselprofil von Krebs therapeutisch auszunutzen. In dieser Studie untersuchten wir die Aktivität und das Zusammenspiel von zwei auf den Stoffwechsel ausgerichteten Medikamenten, Metformin und Dichloracetat (DCA), um ihre Auswirkungen auf Wachstum und Überleben von Brustkrebszellen zu bestimmen.
Metforminhydrochlorid (1,1-Dimethylbiguanidhydrochlorid) ist ein oral einzunehmendes Medikament, das häufig zur Behandlung von Typ-2-Diabetes eingesetzt wird. Studien haben gezeigt, dass bei Diabetikern, die Metformin einnehmen, weniger Krebserkrankungen und damit verbundene Todesfälle auftreten als bei Patienten, die kein Metformin einnehmen. In einer solchen Studie sprachen diabetische Brustkrebspatientinnen, die Metformin einnahmen, deutlich besser auf eine neoadjuvante Chemotherapie an als Patientinnen, die kein Metformin erhielten [6-8]. In-vitro-Studien haben ergeben, dass Metformin das Wachstum vieler Arten von Krebszellen hemmt, einschließlich derjenigen von Brustkrebs, Dickdarmkrebs, Prostatakrebs, Eierstockkrebs und Gliomen [9-12]. Es ist bekannt, dass Metformin die AMP-aktivierte Proteinkinase (AMPK) aktiviert, was zu einer Hemmung der Proteinsynthese und des Zellwachstums führt [13]. Die Aktivierung der AMPK allein reicht jedoch nicht aus, um den apoptotischen Zelltod herbeizuführen [14]. Studien haben gezeigt, dass Metformin sich in den Mitochondrien anreichert und den Komplex I der Elektronentransportkette leicht hemmt, ein Vorgang, der der AMPK-Aktivierung vorgeschaltet ist [15-18]. Da Komplex I gehemmt wird, führt der behinderte Elektronendurchgang zur Superoxidproduktion innerhalb der mitochondrialen Matrix, wodurch mitochondriale Proteine, Lipide und Nukleinsäuren geschädigt werden. In Studien, in denen nachgewiesen wurde, dass Metformin den Zelltod fördert, ist die Apoptose der Hauptweg [10, 12, 19]. Wir haben bereits gezeigt, dass Metformin bei den meisten Brustkrebs-Zelllinien sowohl den Kaspase-abhängigen als auch den Poly(ADP-Ribose)-Polymerase (PARP)-abhängigen Zelltod auslöst, während es für nicht-transformierte Brustepithelzellen nicht zytotoxisch ist [20]. Der Poly(ADP-Ribose)-Polymerase-abhängige Zelltod war mit erheblichen Veränderungen der mitochondrialen Form und Funktion verbunden, was zu der Schlussfolgerung führt, dass mitochondriale Schäden in Krebszellen ein Schlüsselvermittler des Metformin-induzierten Zelltods sind. Auf der Grundlage dieser Beobachtungen stellten wir die Hypothese auf, dass Verbindungen, die den oxidativen Stoffwechsel der Mitochondrien fördern, die Metformin-induzierte mitochondriale Schädigung verstärken und mit Metformin bei der Abtötung von Krebszellen synergistisch wirken würden. Da die Behandlung mit Metformin auch die Produktion von Laktat [21]fördert, würde eine solche Verbindung idealerweise auch diese Wirkung bekämpfen.
Dichloracetat ist ebenfalls ein oral verfügbares Medikament mit gut untersuchter Pharmakokinetik und wurde für die Behandlung von Laktatazidose (eine mögliche Nebenwirkung von Metformin) und mitochondrialen Defiziten getestet [27]. Dichloracetat ist ein Inhibitor der PDK, die die Pyruvatdehydrogenase (PDH) phosphoryliert und dadurch inaktiviert [23]. Pyruvatdehydrogenase ist das Enzym, das die Umwandlung von Pyruvat in Acetyl-CoA katalysiert, das in den mitochondrialen Tricarbonsäurezyklus (TCA) und die oxidative Phosphorylierung gelangt. In Krebszellen ist die PDK-Aktivität häufig erhöht und fungiert als Gatekeeper, der den Fluss von Pyruvat aus dem Zytoplasma in den Mitochondrien-Stoffwechsel reduziert. Man geht davon aus, dass dies eine wichtige Komponente der metabolischen Umprogrammierung in Krebszellen ist, die zu einer verringerten Glukoseoxidation und der Produktion von Laktat führt [24-26]. Durch Hemmung der PDK steigert DCA die PDH-Aktivität, so dass Pyruvat in den TCA-Zyklus gelangen kann, anstatt in Laktat umgewandelt und ausgeschieden zu werden [27].
In dieser Studie untersuchten wir die Antitumoraktivität und das Zusammenspiel von zwei auf den Stoffwechsel ausgerichteten Medikamenten, Metformin und DCA. Wir zeigen, dass DCA die Zytotoxizität von Metformin auf Brustkrebszellen durch einen Mechanismus verstärkt, der oxidative Schäden einbezieht, während es gleichzeitig die Laktatproduktion durch Metformin senkt, was möglicherweise einen doppelten therapeutischen Vorteil bietet.
Methoden
Chemikalien und Reagenzien
Die folgenden Chemikalien, Reagenzien und Kits wurden von Sigma-Aldrich bezogen, sofern nicht anders angegeben: Metformin (1,1-Dimethylbiguanid), Natriumdichloracetat, 0.4 %ige Trypanblau-Lösung, Vectashield-Fluoreszenzmedium mit 4,6-Diamidino-2-Phenylindol (DAPI) (Vector Laboratories), Lactate Assay Kit (Eton Biosciences), caspase-Inhibitor OPH-109 (MP Biomedicals), Coomassie Brilliant Blue R250 (Bio-Rad Laboratories), Paraformaldehyd, SYTOX® Green (Life Technologies), Triton X-100 (Eastman) und PARP-Inhibitor II INH2BP (Epigentek).
Zellkultur
Die menschlichen Brustkrebszelllinien MCF7 und T47D sowie die menschlichen Brustepithelzellen MCF10A wurden von ATCC bezogen. Die 66CL4 Maus-Mammakarzinom-Zelllinie wurde von Dr. Fred Miller (Karmanos Cancer Institute, Detroit, MI) zur Verfügung gestellt. Nach Erhalt der Zelllinien wurden die Zellen sofort kultiviert und expandiert, um gefrorene Ampullenbestände herzustellen. Die Zellen wurden nicht länger als 2-3 Monate passagiert, bevor neue Kulturen aus den gefrorenen Ampullen der ersten Passage angelegt wurden. Die Zelllinien wurden routinemäßig auf Mykoplasmenkontamination überprüft und von IDEXX RADIL Laboratories als mykoplasmenfrei bestätigt. Die Zellen wurden in Dulbecco’s modified Eagle’s medium (DMEM) mit 10 % fötalem Rinderserum, 100 U/ml Penicillin und 100 µg/ml Streptomycin gehalten. Die Zellen wurden in einem befeuchtetenCO2-Inkubator bei 37 °C inkubiert.
Trypanblau-Ausschluss-Assay
MCF7- und T47D-Zelllinien wurden auf 35-mm-Schalen ausgeplattet. Die Zellen wurden mit Metformin und DCA in den angegebenen Konzentrationen oder mit Vehikel behandelt. Nach der angegebenen Zeitspanne wurde das Medium entnommen, um schwimmende tote Zellen zu retten. Anhaftende Zellen wurden mit phosphatgepufferter Kochsalzlösung (PBS) gewaschen, die mit dem gesammelten Medium gepoolt wurde. Anschließend wurden die Zellen durch Trypsinierung geerntet und dem Pool hinzugefügt. Mit Trypanblau-Lösung wurden die toten Zellen im Verhältnis 1:1 angefärbt, und die Zellen wurden mit einem Hämazytometer gezählt.
Western Blotting
Die Zellen wurden auf 35-mm-Schalen ausgebreitet. Nach der Behandlung über den angegebenen Zeitraum wurden die Zellen im Medium geerntet, um lebende und tote Zellen zu sammeln. Die Zellen wurden pelletiert, mit PBS gewaschen und erneut pelletiert. Das Zellpellet wurde dann durch Zugabe von 1× Natriumdodecylsulfat (SDS)-Probenpuffer [2,5 mM Tris-HCl (pH 6,8), 2,5 % SDS, 100 mM Dithiothreitol, 10 % Glycerin, 0,025 % Bromphenolblau] lysiert. Gleiche Proteinmengen wurden auf einem 8,5 %igen SDS-Polyacrylamid-Gel aufgetrennt. Die Proteine wurden mit einer Bio-Rad Trans-Blot-Apparatur unter Verwendung eines Transferpuffers [48 mM Tris-HCl, 39 mM Glycin] auf Immobilon-P-Membranen (Millipore) übertragen. Die Membranen wurden in 5 % fettfreier Trockenmilch in Tris-gepufferter Kochsalzlösung mit Tween 20 (TBS-T) [10 mM Tris-HCl (pH 7,5), 150 mM NaCl, 0,1 % Tween-20] mit dem angegebenen Antikörper entweder 3 Stunden lang bei Raumtemperatur oder über Nacht bei 4 °C eingelegt. Nach gründlichem Waschen mit TBS-T wurde ein geeigneter, mit Meerrettichperoxidase (HRP) konjugierter Sekundärantikörper aufgetragen. Die Membran wurde erneut gewaschen, und die Proteine wurden dann mit dem chemilumineszenten Substrat Super Signal West Pico (Pierce Biochemical) nachgewiesen. Für die Isolierung der Mitochondrien (Abb. 3d) wurden MCF7-Zellen bis zu 95 % Konfluenz auf 150-mm-Schalen gezüchtet. Zur Isolierung der Mitochondrien wurde ein Mitochondrien-Isolierungskit für kultivierte Zellen (Pierce) gemäß dem Protokoll des Herstellers verwendet. Alle Blots wurden mit einem UVP-Imaging-System abgebildet. Es wurden die folgenden Antikörper verwendet: Pyruvatdehydrogenase-Kinase1 (Abcam), PARP (Cell Signaling), Anti-4-Hydroxynonenal (Millipore), Komplex I-Untereinheit NDUFB8 (Mitosciences), GAPDH (Ambion), PDH (Abcam) und Phospho-PDHE1 alpha (Calbiochem).
Konfokale Mikroskopie
Die Zellen wurden auf Glasdeckgläsern in 35-mm-Schalen ausplattiert und wie angegeben behandelt. Die Zellen wurden in PBS gewaschen, 10 Minuten lang mit 4 % Paraformaldehyd fixiert, in Vectashield-Medium mit DAPI auf Standard-Objektträger aufgezogen und dann mit einem konfokalen Mikroskop Olympus FV1000 bei 100× beobachtet. Die Etablierung von MCF7-Zelllinien, die pAcGFP1-Mito (Clontech; ein Plasmid, das für mitochondriale fluoreszierende Proteine kodiert) stabil exprimieren, wurde zuvor beschrieben [20].
Laktattest und pH-Analyse
MCF7- und 66CL4-Zellen in 35-mm-Schalen wurden zu 80 % konfluiert und wie angegeben in phenolrot- und karbonatfreiem Medium behandelt. Die Zellen wurden 4-6 Stunden lang behandelt. Das Medium aus den Schalen wurde gesammelt und die Laktatkonzentration mit einem im Handel erhältlichen L-Laktat-Assay-Kit (Eton Bioscience) gemessen. Bei der 66Cl4-Zelllinie wurden die Zellen zur Normalisierung des Laktatspiegels mit einem Hämatometer gezählt. Der pH-Wert des Mediums wurde mit einem pH-Meter gemessen.
Koloniebildungstest
MCF7-Zellen wurden zu 500 Zellen pro 60-mm-Schale ausgeplattet. Am folgenden Tag wurden die Zellen wie angegeben behandelt. Nach 12 Tagen Inkubation wurden die Zellen mit PBS gewaschen und mit 70 %igem Ethanol für 5 Minuten fixiert. Anschließend wurden die Kolonien mit Coomassie-Blau [40 % Methanol, 12 % Eisessig, 0,24 % Coomassie-Blau] angefärbt, gewaschen, abgebildet und mit einem AlphaImager-System und der entsprechenden Bildanalyse-Software (AlphaInnotech, Santa Clara, CA) quantifiziert.
Durchflusszytometrie
MCF7-Zellen wurden wie angegeben behandelt. Die Zellen wurden dann 15 Minuten lang mit MitoSOX (5 µM) inkubiert, gewaschen, trypsiniert und 1 ml 10 % FBS DMEM zu den Zellen gegeben. Der Nachweis der MitoSOX-Konzentration in einer gleichen Anzahl von Zellen pro Behandlungsbedingung wurde durchflusszytometrisch mit einem Accuri C6-Zytometer bestimmt.
Hemmung von PDK1 mit einer siRNA
On-TARGET Plus Smartpool siRNA, die auf PDK1 abzielt, und nicht abzielende siRNA wurden von Thermo Scientific erworben. 66CL4-Zellen (250.000/Schale) wurden auf 35-mm-Schalen ausplattiert. Am folgenden Tag wurden die Zellen mit den siRNAs unter Verwendung von Dharmafect (Thermo Scientific) gemäß dem Herstellerprotokoll transfiziert. Die Transfektion wurde am darauffolgenden Tag wiederholt. Am folgenden Tag wurden die Zellen gewaschen und wie angegeben in karbonatfreier DMEM mit 10 % FBS behandelt. Nach 4 Stunden wurden die Zellen mit dem Hämacytometer gezählt, und das Medium wurde zur Messung des pH-Werts und des Laktatgehalts entnommen. Die Zellen wurden für die Analyse der PDK1-Konzentration mittels Western Blotting entnommen.
Test auf reaktive Thiobarbitursäure (TBARS)
MCF7-Zellen wurden auf 150-mm-Schalen zu 100 % konfluiert. Die Zellen wurden wie angegeben 24 Stunden lang mit Metformin (8 mM) oder DCA (5 mM) behandelt. Die Zellen wurden in PBS geerntet, beschallt und die relativen Proteinwerte wurden mit dem Bradford-Assay bestimmt. Die Lysate wurden mit dem Quantichrom™ TBARS Assay Kit verarbeitet und auf einem SpectraMax M5-Plattenlesegerät abgelesen(λ ex/em = 560 nm/585 nm).
SYTOX® green assay
MCF7- und T47D-Zellen wurden auf 96-Well-Platten ausplattiert und zu 90 % konfluiert. Die Zellen wurden wie angegeben behandelt und anschließend mit dem Nukleinsäure-Farbstoff SYTOX® Green (10 µM) versetzt und 20 Minuten lang inkubiert, bevor sie mit einem Fluoreszenz-Plattenlesegerät bei λ ex/em = 485/535 nm mit einem Cutoff von 515 nm abgelesen wurden. Die Zellen wurden dann 30 Minuten lang mit Triton X-100 (0,4 %) permeabilisiert, und es wurde eine zweite Messung durchgeführt, um die Gesamtmenge der DNA-Färbung zu bestimmen, die ein Surrogat für die Gesamtzahl der Zellen darstellt. Die Kombinationsindexwerte wurden mit der Software CalcuSyn ermittelt.
Sauerstoffverbrauchsrate
Die Sauerstoffverbrauchsrate(OCR) wurde mit einem Seahorse XF24-Analysegerät gemäß den Anweisungen des Herstellers (Seahorse Bioscience, North Billerica, MA, USA) gemessen. T47D-Zellen wurden mit 40.000 Zellen pro Vertiefung in XF24-Well-Platten plattiert. Am nächsten Tag wurden die Zellen gewaschen und 30 Minuten lang mit pufferfreien Medien bei 37 °C in einemCO2-freien Inkubator äquilibriert, bevor sie in das XF24-Analysegerät übertragen wurden. Nach einer ersten Messung der OCR wurde DCA über Injektionsöffnungen in die Vertiefungen bis zu einer Endkonzentration von 5 mM zugegeben. Die OCR wurde alle 30 Minuten gemessen.
Statistische Analyse
Der Vergleich zweier Gruppen erfolgte mit Hilfe eines ungepaarten t-Tests mit Welch’s Korrektur, der mit der Software Graph Pad Prism (La Jolla, CA) erstellt wurde. P-Werte, die kleiner oder gleich 0,05 waren, wurden als signifikant angesehen.
Ergebnisse
DCA und Metformin induzieren synergistisch Apoptose in Brustkrebszellen
Wir haben bereits gezeigt, dass Metformin nach dreitägiger Behandlung bei den meisten Brustkrebszelllinien zwei verschiedene Arten von Zelltod auslöst, während es für normale Brustepithelzellen nicht zytotoxisch ist [20]. Die Induktion des Zelltods in Brustkrebszellen stand in engem Zusammenhang mit einer veränderten Mitochondrienstruktur, was darauf hindeutet, dass Metformin, ein bekannter Inhibitor des Komplexes I der mitochondrialen Elektronentransportkette, Krebszellen durch eine Veränderung der Mitochondrienfunktion tötet. Dies veranlasste uns zu der Hypothese, dass eine Steigerung des mitochondrialen Elektronentransports den Metformin-induzierten Tod von Krebszellen verstärken würde. Da DCA die mitochondriale Oxidation von Pyruvat fördert, haben wir zunächst untersucht, ob DCA die Metformin-induzierte Zytotoxizität in Brustkrebszelllinien verstärken könnte.
MCF7- und T47D-Brustkrebszellen wurden mit Metformin, DCA oder deren Kombination behandelt. Die verwendete Metformin-Konzentration betrug 8 mM, was einer physiologisch relevanten Metformin-Dosis entspricht, wie die Arbeit von Owen et al. [17]. Wie bei DCA können im Patientenserum niedrige millimolare Konzentrationen erreicht werden, so dass der Bereich von 0,5-5 mM klinisch relevant ist [27-29] und wurde in diesen Experimenten verwendet. Die Anzahl der lebenden und toten Zellen wurde mit dem Trypanblau-Ausschlussverfahren bestimmt (Abb. 1a). Der Zelltod wurde bei MCF7-Zellen (Abb. 1a links) gemessen, die 2 Tage lang behandelt wurden, und bei T47D-Zellen (Abb. 1arechts), die 4 Tage lang behandelt wurden. Bei beiden Zelllinien induzierte Metformin den Zelltod, wie in früheren Experimenten beobachtet [20]. Der Zelltod wurde jedoch durch die gleichzeitige Behandlung mit DCA und Metformin deutlich erhöht, während DCA allein nur minimale zytotoxische Wirkungen hatte (Abb. 1a). Als MCF10A nicht-transformierte Brustepithelzellen den gleichen Behandlungen unterzogen wurden, wurde kein Zelltod beobachtet (Abb. 1b). Um diese Beobachtung des Zelltods in Krebszellen weiter zu untersuchen, wurden Dosis-Titrationen und SYTOX Green Zytotoxizitätstests durchgeführt (Abb. 1c). Die Wirkstoffkombination wurde nach der Methode von Chou auf Synergie getestet [30] mit der Software CalcuSYN auf Synergie geprüft, die einen Kombinationsindex (CI) der Wirkstoffe generiert. MCF7-Zellen (Abb. 1c links) hatten einen CI von 0,00047, wenn sie zwei Tage lang mit der Kombination aus DCA und Metformin behandelt wurden. T47D-Zellen wiesen nach 4 Tagen Behandlung einen KI von 9,762e-006 auf (Abb. 1c rechts). Diese Werte weisen auf eine starke synergistische Zytotoxizität der Kombination von DCA und Metformin bei diesen Brustkrebszelllinien hin.
Anschließend wurden die beobachteten zytotoxischen Wirkungen von DCA (2,5 mM) und Metformin (1 mM) mit Hilfe von Koloniebildungstests validiert. Bei diesen Konzentrationen verringerten beide Wirkstoffe allein die Koloniegröße, aber nicht signifikant die Koloniezahl (Abb. 1d, e). Bei der Kombination der beiden Wirkstoffe wurde jedoch ein signifikanter Rückgang der Koloniezahl beobachtet (Abb. 1e). Diese Beobachtungen zeigen, dass DCA die Metformin-induzierte Zytotoxizität in Brustkrebszellen im Vergleich zu einem der beiden Wirkstoffe allein verstärkt.
DCA fördert die oxidative Phosphorylierung durch Hemmung von PDK1 und dämpft die Metformin-induzierte Laktatproduktion
Um zu bestätigen, dass die beobachteten Wirkungen von DCA auf die Hemmung des erwarteten Ziels, PDK1, zurückzuführen sind, zeigen wir, dass DCA tatsächlich die Menge an phosphorylierter PDH im Vergleich zur Kontrolle in MCF7- und MCF10A-Zellen durch Western Blot verringert (Abb. 2a). Es wird erwartet, dass die Stimulierung der Pyruvatdehydrogenase nach einer DCA-Behandlung den oxidativen Metabolismus von Pyruvat verbessert. Dieser Effekt wurde mit einem Seahorse XF24-Analysegerät untersucht. Es wurden die OCRs von Kontroll- und DCA-behandelten MCF7-Zellen gemessen (Abb. 2b). Mit Dichloracetat behandelte Zellen wiesen im Vergleich zu den Kontrollzellen eine signifikant höhere OCR auf.
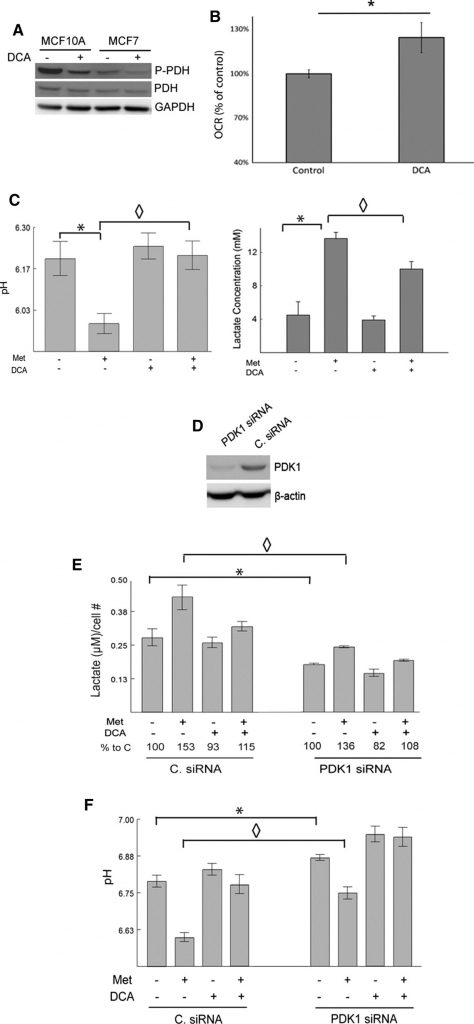
Es wird erwartet, dass Metformin als milder Inhibitor von Komplex I und als Verstärker der AMPK-Aktivität die glykolytischen Raten stimuliert und die Laktatproduktion erhöht [11,14,15,17]. Im Gegensatz dazu wird erwartet, dass DCA Pyruvat auf Kosten der Laktatproduktion in den mitochondrialen Stoffwechsel umlenkt. Um dies zu untersuchen, wurden der pH-Wert und die Laktatkonzentration im Medium kultivierter MCF7-Zellen nach Behandlung mit Metformin, DCA oder beidem gemessen. Mit Metformin behandelte Zellen produzierten signifikant höhere Laktatmengen als unbehandelte Kontrollzellen. Dichloracetat allein hatte nur geringe Auswirkungen, verringerte aber die Metformin-induzierte Laktatproduktion erheblich. Der pH-Wert des Mediums korrespondierte mit den beobachteten Veränderungen des Laktatspiegels (Abb. 2c). Das heißt, Metformin senkte den pH-Wert des Mediums erheblich, und dieser Effekt wurde durch DCA weitgehend aufgehoben.
Um die Rolle von PDK bei der Laktatproduktion und der Hemmung des Eintritts von Pyruvat in den TCA-Zyklus weiter zu untermauern, wurde PDK mit siRNA in 66CL4-Zellen ausgeschaltet. Diese Zelllinie stammt von einem Mammakarzinom der Maus und könnte sich als nützlich erweisen, um diese Experimente in künftigen Studien in eine präklinische Umgebung zu übertragen. Die Zellen wurden entweder mit einer gegen PDK1 gerichteten siRNA (PDK1siRNA) oder mit einer nicht gerichteten Kontroll-siRNA (C. siRNA) transfiziert. Die PDK1-Konzentrationen wurden mittels Western Blot nachgewiesen, und die mit PDK1siRNA transfizierten Zellen wiesen im Vergleich zu den mit C. siRNA transfizierten Zellen deutlich niedrigere PDK1-Konzentrationen auf (Abb. 2d). Die siRNA-vermittelte Unterdrückung von PDK1 reduzierte die Laktatproduktion und erhöhte den pH-Wert des Mediums, was mit den bekannten Funktionen von PDK1 übereinstimmt (Abb. 2e, f). Der Knockdown von PDK1 verringerte effektiv die Metformin-induzierten Veränderungen bei Laktat und dem pH-Wert des Mediums. Dichloracetat verringerte das Laktat weiter und erhöhte den pH-Wert in den mit PDK1 siRNA transfizierten Zellen. Die Fähigkeit von DCA, die Auswirkungen von Metformin auf Laktat und pH-Wert umzukehren, wurde etwas abgeschwächt, wenn PDK1 ausgeschaltet wurde, was wiederum darauf hindeutet, dass DCA seine Wirkungen über PDK1 vermittelt. Zusammenfassend lässt sich sagen, dass der Verlust von PDK1 entweder durch siRNA-Transfektion oder durch Hemmung durch DCA die Metformin-induzierte Laktatproduktion verhindert.
DCA in Kombination mit Metformin verstärkt oxidative Schäden und den anschließenden Caspase-abhängigen Zelltod
Metformin hemmt den Komplex I der Elektronentransportkette, was den Elektronenfluss behindert und zur Produktion von Superoxid innerhalb der mitochondrialen Matrix führt. Da DCA den Eintritt von Pyruvat in den TCA-Zyklus fördert, stellten wir die Hypothese auf, dass DCA die ROS-Produktion als einen möglichen Mechanismus seiner zytotoxischen Synergie mit Metformin verstärken könnte. MitoSOX, ein spezifischer Indikator für die mitochondriale Superoxidproduktion, färbte Zellen an, die mittels Durchflusszytometrie analysiert wurden. Nach einer einstündigen Behandlung erhöhten weder Metformin (rote Linie, Abb. 3a, linkes Feld) noch DCA (nicht gezeigt) die mitochondriale Superoxidproduktion. Die kombinierte Behandlung mit DCA und Metformin führte jedoch zu einer Rechtsverschiebung der Fluoreszenzintensität, was auf eine erhöhte Superoxidproduktion hinweist. Nach 3 Stunden Behandlung begannen mit Metformin behandelte Zellen im Vergleich zu unbehandelten Zellen (schwarze Linie) vermehrt mitochondriales Superoxid zu produzieren (rote Linie, Abb. 3a, rechtes Feld). Die Kombination von DCA und Metformin führte zu einer noch stärkeren Superoxidfärbung. Diese Ergebnisse deuten darauf hin, dass Metformin den Elektronentransport im Komplex I beeinträchtigt, was zu einer erhöhten Superoxidproduktion führt, und dass DCA die Wirkung von Metformin potenziert.
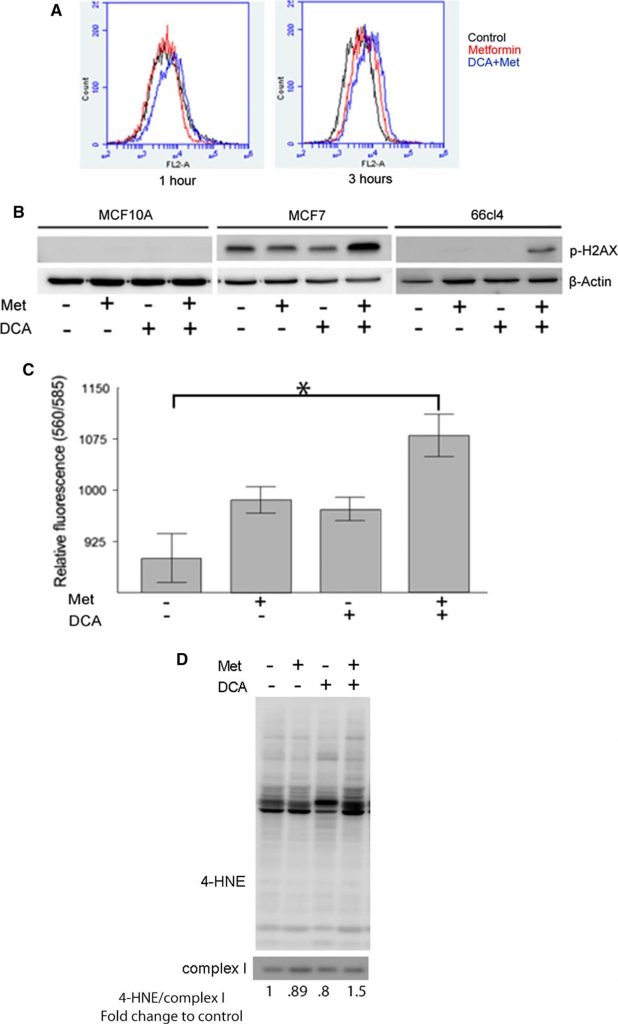
Die Anhäufung von Superoxid und anderen ROS hat das Potenzial, zelluläre Komponenten wie Proteine, Membranen und DNA zu schädigen. Oxidative DNA-Schäden können zu Doppelstrangbrüchen und zur Phosphorylierung von Histon H2AX (p-H2AX) führen, das ein nützlicher Surrogatmarker für solche DNA-Schäden ist [31]. Um die mit der ROS-Produktion verbundenen DNA-Schäden als Reaktion auf die Behandlung mit Metformin und DCA abzuschätzen, kann daher der Gehalt an phosphoryliertem H2AX analysiert werden. Menschliches Brustepithel MCF10A und die Brustkrebszelllinien MCF7 und 66CL4 wurden nach einer 24-stündigen Behandlung mit DCA, Metformin oder deren Kombination mittels Western Blotting analysiert (Abb. 3b). In MCF10A-Zellen wurden bei keiner der Behandlungen nachweisbare Mengen an p-H2AX beobachtet. In MCF7-Zellen war p-H2AX in unbehandelten Zellen vorhanden und wurde durch die Kombination von Metformin und DCA beträchtlich erhöht, aber nicht durch eines der beiden Medikamente allein. In 66CL4-Zellen wurde p-H2AX nur in DCA- und Metformin-behandelten Zellen beobachtet. Diese Daten deuten darauf hin, dass DCA und Metformin zusammen die oxidative Schädigung der DNA verstärken.
Als zweites Beweismittel haben wir die Lipidperoxidation gemessen, die auftritt, wenn freie Radikale die Lipide in den Zellmembranen schädigen. Zur Abschätzung des Ausmaßes der Lipidoxidation nach der Behandlung mit DCA und Metformin wurden zwei verschiedene Methoden verwendet. Erstens der TBARS-Test, der Nebenprodukte der Lipidperoxidation misst und zur Bewertung des Ausmaßes der durch ROS verursachten oxidativen Schädigung von Zellmembranen verwendet wird. TBARS stieg in MCF7-Zellen an, die entweder mit Metformin oder DCA allein behandelt wurden, und der stärkste Anstieg wurde beobachtet, wenn beide Medikamente kombiniert wurden (Abb. 3c). Die zweite Methode zur Abschätzung der Lipidoxidation war ein Western Blot von 4-Hydroxynonenol (4-HNE)-Proteinaddukten, die aus der Oxidation von ungesättigten Fettsäuren in Zellmembranen stammen und kovalente Addukte mit Proteinen bilden, was einen Hinweis auf oxidative Schäden an Zellmembranen liefert. MCF7-Mitochondrienextrakte von Zellen, die 24 Stunden lang mit DCA, Metformin oder beidem behandelt wurden, wurden auf den Gehalt an 4-HNE-Proteinaddukten untersucht (Abb. 3d). Erhöhte Gehalte an mitochondrialen 4-HNE-Addukten wurden nur in Zellen beobachtet, die mit der Kombination aus Metformin und DCA behandelt wurden. Dies bestätigt frühere Ergebnisse, wonach die Kombination dieser Medikamente eine erhöhte Produktion von ROS verursacht.
Unsere bisherigen Daten zeigen, dass metforminempfindliche Zellen große Vakuolen entwickeln, die durch die Darstellung von GFP-verknüpften Mitochondrien und durch Elektronenmikroskopie als strukturell veränderte Mitochondrien bestimmt wurden. Diese veränderten Mitochondrien scheinen spezifisch mit einem PARP-abhängigen Zelltodweg verbunden zu sein, der im Vergleich zum apoptotischen Zelltod zeitlich verzögert ist [20]. Da DCA beim Zelltod mit Metformin synergiert, wurden die Auswirkungen von DCA auf die mitochondriale Struktur untersucht. Es wurde eine stabile MCF7-Zelllinie entwickelt, die ein auf Mitochondrien ausgerichtetes grün fluoreszierendes Protein exprimiert (pAcGFP1-Mito). Die Zellen wurden einen Tag lang behandelt, bevor sie mit Phasenkontrast- und konfokaler Mikroskopie untersucht wurden (Abb. 4). In Zellen, die nur mit Metformin behandelt wurden, wurden große Mitochondrien beobachtet, wie bereits zuvor gesehen [20]. Im krassen Gegensatz dazu wiesen DCA-behandelte Zellen kleinere und zahlreichere Mitochondrien auf. Dichloracetat verhinderte auch die durch Metformin verursachte Vergrößerung der Mitochondrien. Im Phasenkontrast waren die vergrößerten Mitochondrien in den mit Metformin behandelten Zellen sichtbar, während sie in der Kombinationsbehandlung fehlten. Eine zweite metforminempfindliche Zelllinie, T47D, wurde ebenfalls untersucht. Auch hier wurden bei der Behandlung mit Metformin vergrößerte Mitochondrien beobachtet, während diese bei der Kombinationsbehandlung fehlten.
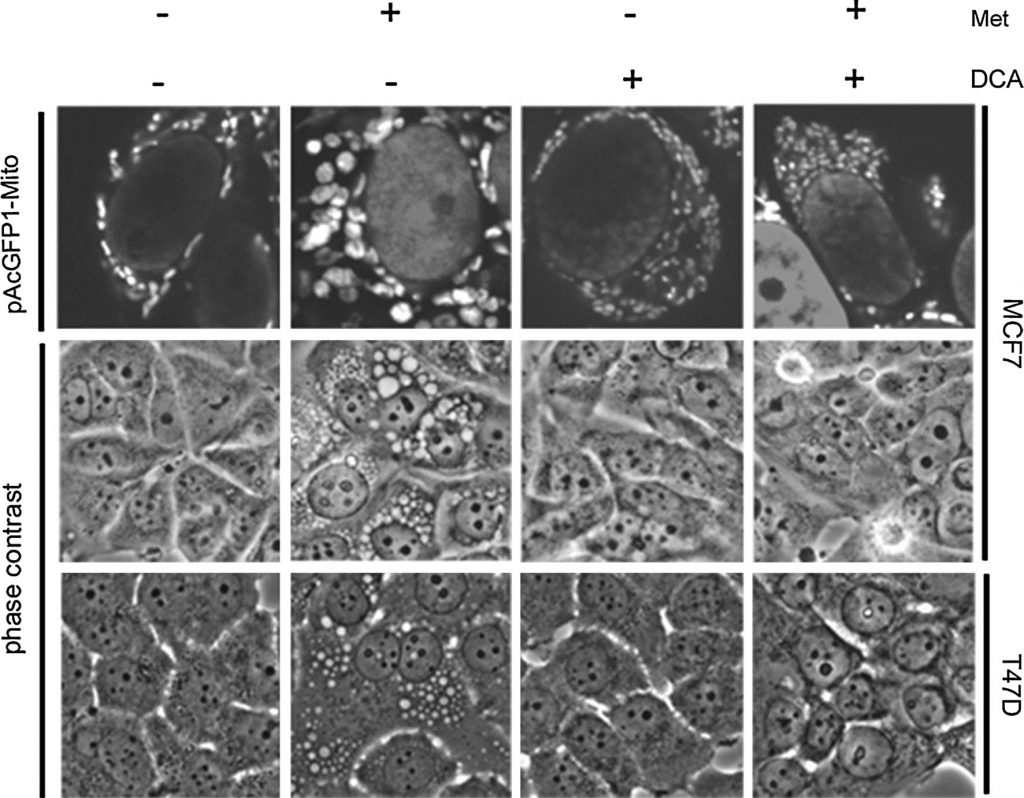
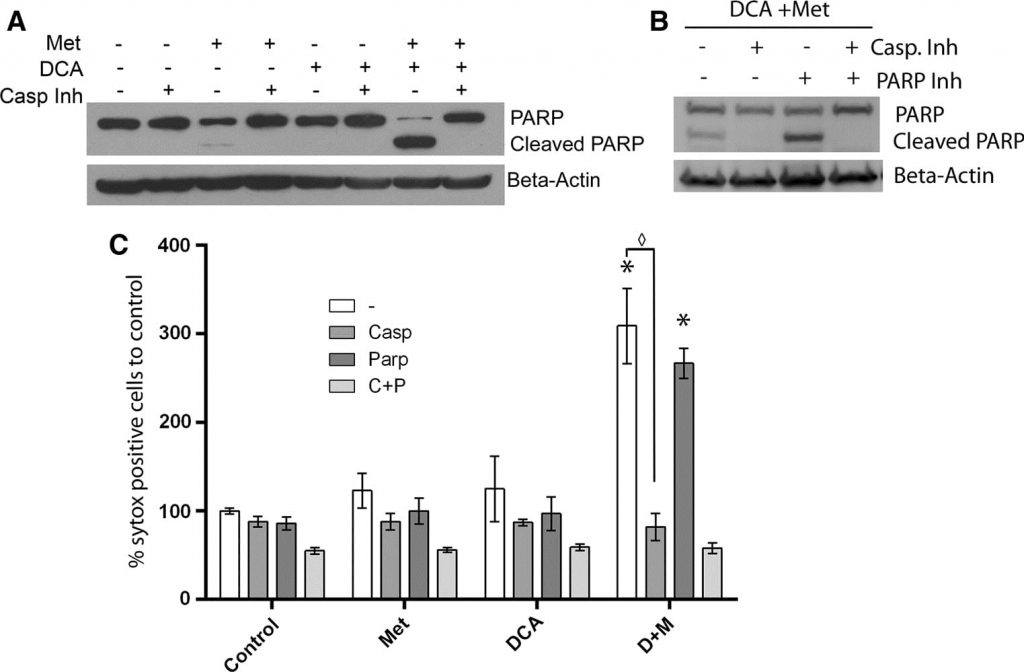
In unserer früheren Arbeit stellten wir fest, dass die Vergrößerung der Mitochondrien mit dem PARP-abhängigen Zelltod korrelierte, der im Vergleich zum apoptotischen Zelltod zeitlich verzögert war. Dies veranlasste uns zu einer Untersuchung der Besonderheiten des Zelltodmechanismus, der durch DCA in Kombination mit Metformin [20]da keine mitochondriale Vergrößerung beobachtet wird. Um den Zelltod in Zellen zu charakterisieren, die gleichzeitig mit Metformin und DCA behandelt wurden, wurden MCF7-Zellen einen Tag lang mit einem irreversiblen Breitspektrum-Caspaseinhibitor (Q-Val-Asp-OPh) zusammen mit DCA, Metformin oder beiden behandelt. Western Blotting (Abb. 5a) zeigte einen starken Anstieg von gespaltenem PARP und einen Rückgang von intaktem PARP in den mit DCA und Metformin behandelten Zellen. Die Zugabe des Caspase-Inhibitors blockierte die Spaltung von PARP vollständig. Ein ähnlicher Effekt wurde zuvor bei Zellen beobachtet, die 2,5 Tage lang mit Metformin allein behandelt wurden [20]. Zum Zeitpunkt von einem Tag induziert Metformin allein jedoch nur einen leichten Anstieg der gespaltenen PARP. Daher deuten diese Daten darauf hin, dass die kaspaseabhängige Apoptose durch die Zugabe von DCA zu Metformin in diesen Brustkrebszellen beschleunigt wird. Um weiter zu untersuchen, ob der PARP-abhängige Tod durch die gleichzeitige Behandlung mit DCA und Metformin eintritt, wurde auch ein PARP-Inhibitor verwendet. Zwei Tage nach der Behandlung wurde zur Quantifizierung des Zelltods ein SYTOX-Green-Assay durchgeführt (Abb. 5c), um festzustellen, ob der PARP-Inhibitor eine zusätzliche Wirkung zum Caspase-Inhibitor hat. Der PARP-Inhibitor hatte weder allein noch in Kombination mit dem Caspase-Inhibitor eine schützende Wirkung. Ein Western Blot bestätigte diesen Befund (Abb. 5b) und zeigte, dass der PARP-Inhibitor die PARP-Spaltung nicht verhinderte. Da die Vergrößerung der Mitochondrien mit dem PARP-abhängigen Zelltod in mit Metformin behandelten Zellen einhergeht, deutet die fehlende Vergrößerung der Mitochondrien bei Zugabe von DCA darauf hin, dass die Apoptose ausschließlich auf kaspaseabhängige Weise stattfindet.
Diskussion
Metformin ist ein seit langem bekanntes Medikament, das zur Behandlung von Typ-2-Diabetes eingesetzt wird. Ebenso wurde DCA in klinischen Versuchen zur Behandlung von Laktatazidose und mitochondrialer Defizite untersucht. Metformin hat bereits gezeigt, dass es die Proliferation vieler Krebszelllinien verlangsamt und deren Zelltod durch Apoptose fördert [9, 10, 12, 32]. Metformin konzentriert sich in den Mitochondrien, wo es den Komplex I der Elektronentransportkette hemmt, was zu einem abnormalen Fluss von Elektronen zu Sauerstoff führt und Superoxid in der Mitochondrienmatrix erzeugt [22, Abb. 3]. Die Produktion reaktiver Sauerstoffspezies ist für Brustkrebszellen problematisch, da viele von ihnen eine geringere Expression von Mangan-Superoxiddismutase (MnSOD) aufweisen, die normalerweise mitochondriales Superoxid eliminiert [33]. Während die pro-apoptotischen und wachstumshemmenden Wirkungen von Metformin auf Krebs bemerkenswert sind, könnte unsere Beobachtung, dass Metformin die Glykolyse steigert, was durch eine erhöhte Laktatproduktion in vitro belegt wird [21]zeigt, könnte seine Wirksamkeit als Einzelwirkstoff einschränken. Da hohe Laktatwerte als tumorfördernd beschrieben wurden und mit einem schlechten klinischen Ergebnis verbunden sind [26, 34-38]haben wir versucht, die Metformin-induzierte Laktatproduktion abzuschwächen, um so möglicherweise die therapeutische Wirksamkeit zu verbessern. In dieser Studie fanden wir heraus, dass DCA dazu beiträgt, die durch Metformin induzierte Laktatproduktion abzuschwächen, und dass DCA dies durch Hemmung von PDK1 erreicht.
Unsere Daten zeigen, dass DCA und Metformin synergistisch die Apoptose stärker auslösen als eine alleinige Behandlung mit Metformin. Unsere Daten zeigen auch, dass DCA Pyruvat zur Verwendung in der oxidativen Phosphorylierung umleitet, was im Zusammenhang mit der Komplex-I-Hemmung durch Metformin zu einer Akkumulation von ROS führt. Wir schließen daraus, dass dieser Anstieg der ROS-Produktion zu den schnelleren Apoptoseraten führt.
In unserer früheren Arbeit haben wir gezeigt, dass Metformin nicht nur den kaspase-abhängigen apoptotischen Zelltod auslöst, sondern auch einen später eintretenden PARP-abhängigen Zelltod, der bei Verwendung eines Pan-Kaspase-Inhibitors aufgedeckt wurde [20]. Diese Form des Zelltods war mit morphologischen Veränderungen in den Mitochondrien verbunden. In dieser Studie verhinderte DCA die morphologischen Veränderungen in den Mitochondrien, die sonst durch Metformin ausgelöst werden, während die gleichzeitige Behandlung zu einem schnelleren Zelltod führte. Darüber hinaus verhinderte die Pan-Caspase-Hemmung den Zelltod fast vollständig und eliminierte die PARP-Spaltung, die mit der gleichzeitigen Behandlung von Metformin und DCA einherging. Zusammengenommen deutet dies darauf hin, dass der Zelltodmechanismus, der durch die gleichzeitige Behandlung ausgelöst wird, die kaspaseabhängige Apoptose ist, und dass die Apoptose im Vergleich zu Metformin allein beschleunigt wird. Es ist möglich, dass dieser apoptotische Prozess zu katastrophal ist und keine Zeit für die Vergrößerung der Mitochondrien benötigt, die mit dem PARP-abhängigen Zelltod von Metformin allein einhergeht.
Während der Vorbereitung dieses Manuskripts wurde eine Studie von Choi und Lim [39] veröffentlicht, die die gleichzeitige Behandlung von DCA und Metformin in Krebszellen untersuchte. Unsere Ergebnisse bestätigen und erweitern die Erkenntnisse dieser Studie erheblich. Während sich die Studie von Choi und Lim fast ausschließlich auf HeLa-Zellen konzentrierte, zeigt unsere Studie die Wirksamkeit der Kombination von DCA und Metformin bei mehreren Brustkrebszelllinien. Wichtig ist, dass wir auch zeigen, dass diese Medikamentenkombination die nicht-transformierten Epithelzellen der Brust, MCF10A, unter den gleichen Bedingungen, unter denen die Medikamente Krebszellen abtöten, nicht abtötet. Wir haben sorgfältige Titrationen durchgeführt, die einen sehr starken Synergismus der Medikamente bei der Herbeiführung des Zelltods in Brustkrebszellen belegen. Diese Ergebnisse deuten darauf hin, dass DCA in Konzentrationen wirksam ist, die therapeutisch beim Menschen erreicht werden können [27, 29]und zwar in weitaus niedrigeren Konzentrationen als in den Experimenten der Studie von Choi und Lim (10-20 mM) [39]. Idealerweise wäre die Höchstdosis von DCA zur Behandlung von Krebszellen so niedrig, dass unerwünschte Wirkungen wie Neuropathie vermieden werden. In einer Studie von Michelakis et al. wurden bei Glioblastom-Patienten, die zweimal täglich 6,25 mg/kg oral einnahmen, DCA-Trog-Serumspiegel von 0,5 mM erreicht [28]. Höhere Dosen wie 25 mg/kg werden mit dosisabhängiger Neuropathie in Verbindung gebracht. Folglich dürften die klinisch relevanten Konzentrationen im Bereich von 0,5-5 mM liegen.
Sowohl die hier vorgestellten Ergebnisse als auch die von Choi und Lim [39] zeigten, dass der durch die Kombination von DCA und Metformin verursachte Zelltod mit oxidativem Stress verbunden ist. Wir zeigen außerdem, dass dies mit der Metformin-induzierten Superoxidproduktion in den Mitochondrien zusammenhängt. Dichloracetat erhöhte die mitochondriale Superoxidproduktion in Gegenwart von Metformin weiter, was zu oxidativen Schäden an Mitochondrien, zellulären Lipiden und DNA führte. Neuartige Kombinationen wie Metformin und DCA sind vielversprechend für die Erweiterung der Brustkrebstherapie.
Danksagungen
Dieser Artikel wurde durch Zuschüsse von Susan G. Komen for the Cure (KG100497) und dem National Cancer Institute der National Institutes of Health (1R01CA180033) unterstützt. Die Zentren für Durchflusszytometrie und Bildgebung wurden durch die COBRE-Zuschüsse P20GM103548 und P20GM103620 des National Institute of General Medical Sciences an den National Institutes of Health unterstützt.
Interessenkonflikt
Die Autoren erklären, dass sie sich in keinem Interessenkonflikt befinden.
Ethische Standards
Die Autoren erklären, dass die Experimente mit den geltenden Gesetzen des Landes, in dem sie durchgeführt wurden, übereinstimmen.
REFERENZEN
1 Warburg O (1956) Über den Ursprung von Krebszellen. Wissenschaft 123(80):309-314. doi:10.1016/S0306-9877(96)90136-X
2 DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB (2008) The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7:11-20. doi:10.1016/j.cmet.2007.10.002
3 Zamzami N, Kroemer G (2001) Das Mitochondrium in der Apoptose: wie sich die Büchse der Pandora öffnet. Nat Rev Mol Cell Biol 2:67-71. doi:10.1038/35048073
4 Gottfried E, Kunz-Schughart LA, Ebner S et al (2006) Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 107:2013-2021. doi:10.1182/blood-2005-05-1795
5 Fischer K, Hoffmann P, Voelkl S et al (2007) Hemmende Wirkung von aus Tumorzellen stammender Milchsäure auf menschliche T-Zellen. Blood 109:3812-3819. doi:10.1182/blood-2006-07-035972
6 Jiralerspong S, Palla SL, Giordano SH et al (2009) Metformin und pathologisches vollständiges Ansprechen auf eine neoadjuvante Chemotherapie bei diabetischen Patienten mit Brustkrebs. J Clin Oncol 27:3297-3302. doi:10.1200/JCO.2009.19.6410
7 Evans JMM, Donnelly LA, Emslie-Smith AM et al (2005) Metformin und verringertes Krebsrisiko bei Diabetikern. BMJ 330:1303-1304. doi:10.1136/bmj.38393.572188.EB
8 Libby G, Donnelly LA, Donnan PT et al (2009) New users of Metformin are at low risk of incident cancer. Diabetes Care 32:1620-1625. doi:10.2337/dc08-2175
9 Zakikhani M, Dowling R, Fantus IG et al (2006) Metformin ist ein AMP-Kinase-abhängiger Wachstumshemmer für Brustkrebszellen. Cancer Res 66:10269-10273. doi:10.1158/0008-5472.CAN-06-1500
10 Isakovic A, Harhaji L, Stevanovic D et al (2007) Doppelte Antigliom-Wirkung von Metformin: Zellzyklus-Stillstand und Mitochondrien-abhängige Apoptose. Cell Mol Life Sci 64:1290-1302. doi:10.1007/s00018-007-7080-4
11 Hadad SM, Appleyard V, Thompson AM (2008) Therapeutische Metformin/AMPK-Aktivierung fördert den angiogenen Phänotyp im ERα-negativen MDA-MB-435 Brustkrebsmodell. Breast Cancer Res Treat 114:391. doi:10.1007/s10549-008-0016-3
12 Buzzai M, Jones RG, Amaravadi RK et al (2007) Systemische Behandlung mit dem Antidiabetikum Metformin beeinträchtigt selektiv das Wachstum von Tumorzellen mit p53-Mangel. Cancer Res 67:6745-6752. doi:10.1158/0008-5472.CAN-06-4447
13 Sarbassov DD, Ali SM, Sabatini DM (2005) Wachsende Rolle des mTOR-Signalwegs. Curr Opin Cell Biol 17:596-603. doi:10.1016/j.ceb.2005.09.009
14 Zhuang Y, Miskimins WK (2008) Cell cycle arrest in Metformin treated breast cancer cells involves activation of AMPK, downregulation of cyclin D1, and requires p27Kip1 or p21Cip1. J Mol Signal 3:1-11. doi:10.1186/1750-2187-3-18
15 Hinke SA, Martens GA, Cai Y et al (2007) Methylsuccinat wirkt der Biguanid-induzierten AMPK-Aktivierung und dem Tod von Beta-Zellen der Bauchspeicheldrüse durch Wiederherstellung des mitochondrialen Elektronentransfers entgegen. Br J Pharmacol 150:1031-1043. doi:10.1038/sj.bjp.0707189
16 Zou M-H, Kirkpatrick SS, Davis BJ et al (2004) Activation of the AMP-activated protein kinase by the anti-diabetic drug metformin in vivo. Rolle der mitochondrialen reaktiven Stickstoffspezies. J Biol Chem 279:43940-43951. doi:10.1074/jbc.M404421200
17 Owen MR, Doran E, Halestrap AP (2000) Beweise, dass Metformin seine antidiabetischen Wirkungen durch Hemmung von Komplex 1 der mitochondrialen Atmungskette ausübt. Biochem J 614:607-614. doi:10.1042/0264-6021:3480607
18 Carvalho C, Correia S, Santos MS et al (2008) Metformin fördert die Beeinträchtigung isolierter Rattenleber-Mitochondrien. Mol Cell Biochem 308:75-83. doi:10.1007/s11010-007-9614-3
19d Kefas BA, Cai Y, Kerckhofs K et al (2004) Metformin-induzierte Stimulation der AMP-aktivierten Proteinkinase in Beta-Zellen beeinträchtigt deren Glukoseempfindlichkeit und kann zu Apoptose führen. Biochem Pharmacol 68:409-416. doi:10.1016/j.bcp.2004.04.003
20 Zhuang Y, Miskimins WK (2012) Metformin induziert sowohl den Caspase-abhängigen als auch den Poly(ADP-ribose)-Polymerase-abhängigen Zelltod in Brustkrebszellen. Mol Cancer Res 9:603-615. doi:10.1158/1541-7786.MCR-10-0343.Metformin
21 Bailey CJ, Wilcock C, Day C (1992) Wirkung von Metformin auf den Glukosestoffwechsel im splanchnischen Bett. Br J Pharmacol 105:1009-1013
22StacpoolePW, Lorenz AC, Thomas RG, Harman EM (1988) Dichloracetat bei der Behandlung der Laktatazidose. Ann Intern Med 108:58-63. doi:10.1056/NEJM198308183090702
23 Whitehouse BSUE, Cooper RH, Randle PJ (1974) Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem J 141:761-774. doi:10.1056/NEJM198308183090702
24 Hussien R, Brooks GA (2011) Mitochondrial and plasma membrane lactate transporter and lactate dehydrogenase isoform expression in breast cancer cell lines. Physiol Genomics 43:255-264. doi:10.1152/physiolgenomics.00177.2010
25 Kim J, Tchernyshyov I, Semenza GL, Dang CV (2006) HIF-1-vermittelte Expression von Pyruvatdehydrogenase-Kinase: ein metabolischer Schalter, der für die zelluläre Anpassung an Hypoxie erforderlich ist. Cell Metab 3:177-185. doi:10.1016/j.cmet.2006.02.002
26 Wigfield SM, Winter SC, Giatromanolaki A et al (2008) PDK-1 reguliert die Laktatproduktion bei Hypoxie und ist mit einer schlechten Prognose bei Plattenepithelkarzinomen des Kopfes und Halses verbunden. Br J Cancer 98:1975-1984. doi:10.1038/sj.bjc.6604356
27 Stacpoole PW, Kurtz TL, Han Z, Langaee T (2008) Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv Drug Deliv Rev 60:1478-1487. doi:10.1016/j.addr.2008.02.014
28 Michelakis ED, Sutendra G, Dromparis P et al (2010) Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2:31ra34. doi:10.1126/scitranslmed.3000677
29 Mori M, Yamagata T, Goto T et al (2004) Dichloracetat-Behandlung bei mitochondrialer Zytopathie: Langzeiteffekte bei MELAS. Brain Develop 26:453-458. doi:10.1016/j.braindev.2003.12.009
30 Chou T (2010) drug combination studies and their synergy quantification using the Chou-Talalay Method drug combination studies and their synergy quantification using the Chou-Talalay Method. Cancer Res 70:440-446. doi:10.1158/0008-5472.CAN-09-1947
31 Mah L, Karagiannis TC (2010) γH2AX: ein empfindlicher molekularer Marker für DNA-Schäden und -Reparatur. Leukemia 24:679-686. doi:10.1038/leu.2010.6
32 Alimova IN, Liu B, Fan Z et al (2009) Metformin hemmt das Wachstum von Brustkrebszellen, die Koloniebildung und induziert einen Zellzyklus-Stillstand in vitro. Cell Cycle 8:909-915. doi:7933 [pii]
33 Soini Y, Vakkala M, Kahlos K et al (2001) Die MnSOD-Expression ist in Tumorzellen invasiver Mammakarzinome seltener als in In-situ-Karzinomen oder nicht-neoplastischen Brustepithelzellen. J Pathol 195:156-162. doi:10.1002/path.946
34 Walenta S, Wetterling M, Lehrke M et al (2000) High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers high lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical. Krebsforschung 60:916-921
35 Leek R, Harris AL (2009) Die Expression von Laktatdehydrogenase 5 in Plattenepithelkarzinomen des Kopfes und Halses steht in Zusammenhang mit der Prognose nach radikaler oder postoperativer Strahlentherapie. Onkologie 77:285-292. doi:10.1159/000259260
36 Isidoro A, Casado E, Redondo A et al (2005) Brustkarzinome erfüllen die Warburg-Hypothese und liefern metabolische Marker für die Krebsprognose. Carcinogenesis 26:2095-2104. doi:10.1093/carcin/bgi188
37 Isidoro A, Martínez M, Fernández PL et al (2004) Die Veränderung des bioenergetischen Phänotyps der Mitochondrien ist ein Kennzeichen von Brust-, Magen-, Lungen- und Speiseröhrenkrebs. Biochem J 378:17-20. doi:10.1042/BJ20031541
38 Walenta S, Schroeder T (2004) Laktat in soliden bösartigen Tumoren: mögliche Grundlage einer metabolischen Klassifizierung in der klinischen Onkologie. Curr Med Chem 11:2195-2204
39 Choi YW, Lim IK (2014) Sensitization of metformin-cytotoxicity by dichloroacetate via reprogramming glucose metabolism in cancer cells. Cancer Lett 346:300-308. doi:10.1016/j.canlet.2014.01.015
Verwandte Inhalte: