Jingtao Tong, Ganfeng Xie, Jinxia He, Jianjun Li, Feng Pan, and Houjie Liang
Department of Oncology, Southwest Hospital, Third Military Medical University, 29 Gaotanyan Street, Chongqing 400038, China
Correspondence should be addressed to Houjie Liang, [email protected] Received 27 May 2010; Revised 29 December 2010; Accepted 13 January 2011
Academic Editor: Miguel A. Andrade
Copyright © 2011 Jingtao Tong et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium provided the original work is properly cited.
Dichloroacetate (DCA), an inhibitor of pyruvate dehydrogenase kinase (PDK), has been recently demonstrated as a promising nontoxic antineoplastic agent that promotes apoptosis of cancer cells. In the present study, we aimed to investigate the antitumor effect of DCA combined with 5-Fluorouracil (5-FU) on colorectal cancer (CRC) cells. Four human CRC cell lines were treated with DCA or 5-FU, or a combination of DCA and 5-FU. The cell viability was determined by 3-(4,5-dimethylthiazol-2-yl)- 2,5-diphenyltetrazolium bromide assay. The interaction between DCA and 5-FU was evaluated by the median effect principle. Immunocytochemistry with bromodeoxyuridine (BrdU) was carried out to determine the proliferation of CRC cells. Cell cycle and apoptosis were measured by flow cytometry, and the expression of apoptosis-related molecules was assessed by western blot. Our results demonstrated that DCA inhibited the viability of CRC cells and had synergistic antiproliferation in combination with 5-FU. Moreover, compared with 5-FU alone, the apoptosis of CRC cells treated with DCA and 5-FU was enhanced and demonstrated with the changes of Bcl-2, Bax, and caspase-3 proteins. Our results suggest that DCA has a synergistic antitumor effect with 5-FU on CRC cell lines in vitro.
1. Introduction
Colorectal cancer is one of the most common malignancies worldwide [1]. Other than surgery, treatment for CRC patients relies primarily on chemotherapy, especially the patients with advanced CRC. Among the chemotherapeutic agents for CRC, 5-Fluorouracil (5-FU), which is a classical chemotherapy agent, has been the first line regimen for treating CRC over several decades [2, 3]. However, 5-FU alone is poorly selective to tumor as well as highly toxic to bone marrow, gastrointestinal tract, and skin when used at the therapeutic dose [4].
Metabolic abnormity is one of the critical hallmarks of cancer [5]. As early as the 1920s, Otto Warburg observed that cancer cells generally use glycolysis rather than oxidative phosphorylation for energy [6]. Thus, the metabolic switch to anaerobic respiration through glycolysis from pyruvate, rather than pyruvate conversion to acetyl-CoA by the action of pyruvate dehydrogenase (PDH) in aerobic glucose metabolism, becomes a preferential phenotype of cancer progress. PDH can be inactivated by pyruvate dehydrogenase kinase (PDK) in many glycolytic phenotypes including cancer, while inhibition of PDK switches metabolism to aerobic oxidation which is proved to be disadvantageous to tumour growth [7].
Dichloroacetate (DCA) is a prototypical inhibitor of mitochondrial PDK. By blocking the enzyme, DCA decreases lactate production by shifting the metabolism of pyruvate from glycolysis towards oxidation in the mitochondria. This property has led to trials of DCA for the treatment of lactic acid accumulation disorders [8]. Recently, studies have demonstrated that DCA suppresses tumour growth via inhibition of PDK [9–11]. Michelakis and his colleagues found that DCA restored mitochondrial function, thus restoring apoptosis, killing cancer cells in vitro, and shrinking the tumours in the rats [12].
Combination chemotherapy has been used widely. 5-FU is usually combined with other antineoplastic agents and radiation to enhance its antitumor effect. The clinical insufficiency appears to be caused by the resistance of 5-FU and severe side effects. The strong and selective induction of apoptosis suggests that the PDK inhibitor DCA may potentiate the inhibitory effect of anticancer drugs, thus exceeding the efficacy of current treatment. Herein, we aimed to examine the combined antitumor effects of DCA with 5-FU on the CRC cells, in hoping of assessing a relatively effective and safe regimen potentiated for CRC treatment.
2. Material and Methods
2.1. Cells and Regents. The human colon cancer cell lines LS174T, LoVo, SW620, and HT29 were purchased from American Type Culture Collection (Manassas, VA, USA). Cell culture reagents were purchased from Gibco-Invitrogen (Carlsbad, CA, USA). Cell lines were maintained in Dulbecco’s Modified Eagle’s medium or Leibovitz L-15 medium containing 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin in a 37◦C, 5% CO2 humidified incubator. 5-FU and DCA were purchased from Sigma-Aldrich Co. Ltd. (St. Louis, MO, USA), dissolved in deionized water to make 1 mol/L working solution, filter sterilized, and subsequently diluted in growth medium for treatment.
2.2. Cell Viability Assay. Cell viability was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Sigma-Aldrich). Cells were seeded into 96-well plates (5 × 104 cells per well) and incubated under standard growth conditions overnight from 60% to 70% confluence. Cells were then treated with DCA alone (final concentration 0–90 mM) or with DCA combined with 5-FU (5–200 μM). After a 48-hour treatment, cells were incubated at 37◦C for another 4 hours with MTT (20 μL per well) and the absorbance at 490 nm was measured in a BioRad Model 550 plate reader (Hercules, CA, USA).
2.3. Analysis of Drug Interaction. The interaction between DCA and 5-FU was analyzed by using the median effect principle described by Chou and Talalay [13, 14]. The program enables calculation of combination indices (CIs) which, when smaller than 1, equal to 1, or greater than 1, indicate synergism, additivity, or antagonism, respectively, between two drugs. CIs were calculated by where (Dx)1 and (Dx)2 are the concentrations of DCA alone or 5-FU alone, giving x% growth inhibition, and (D)1 and (D)2 are the drug concentrations in combination inhibiting cell growth also x%.
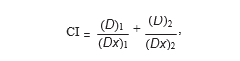
(Dx)1 and (Dx)2 were calculated by the median-effect equation:
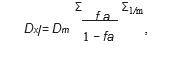
where Dm is the median-effect dose, fa is the fraction affected, and m represents the slope of the median-effect plot.
2.4. Cell Proliferation Assays. Immunocytochemistry was carried out with bromodeoxyuridine (BrdU) (BD Bioscience, San Jose, CA, USA) in vitro. Cells were propagated on coverslips in 12-well plates under standard growth conditions. After 24 hours, various concentrations of DCA, 5-FU, or a combination of two drugs were added. Cells were serum-starved for 12 hours in growth media containing 0.5% FBS to reset the cell cycle to G0 phase, and then cells were pulsed for 2 hours with 10 μmol/L BrdU in growth media. Subsequently, the cells were fixed, washed, and stained according to the manufacturer’s instruction.
Cell cycle analysis was determined indirectly using propidium iodide (PI, BD Bioscience) staining by flow cytometry (FACScan, Becton Dickinson, San Jose, CA, USA). Cells were seeded into 6-well plates and cultured with or without 10 mM DCA or 20 μM 5-FU. After incubating for 48 hours, cells were harvested with 0.25% Trypsin-EDTA (Invitrogen, Carlsbad, CA, USA). Then, cells were fixed with 70% alcohol for 24 hours at 4◦C and washed twice with phosphate buffer saline (PBS). RNAase (100 μL; 1 mg/mL) (BD Bioscience) was added, and cells were incubated in a 37◦C water bath for 30 minutes. After staining with 200 μL PI (50 μg/mL), cells were held at 4◦C for 30 minutes. Finally, cells were analyzed by flow cytometry.
2.5. Apoptosis Assay. Apoptosis was detected by flow cytometry with annexin-V-FITC (BD Bioscience) and PI. Cells were seeded into 6-well plates. After a 48-hour incubation with or without drugs, cells were washed and resuspended in 0.5 mL PBS buffer. After staining with annexin-V-FITC and PI, cells were analyzed by flow cytometry in three independent experiments.
2.6. Western Blot. Cells were harvested and total proteins were extracted with RIPA buffer containing protease inhibitors. Total protein (50 μg) was subjected to 10% or 12% SDS/PAGE, and the resolved proteins were transferred electrophoretically to PVDF membranes (Millipore, Bedford, MA, USA). Membranes were blocked for 2 hours with 5% nonfat milk in TBS buffer containing 0.05% Tween- 20 (TBST) at 4◦C. Membranes were then incubated with antibodies to Bax, Bcl-2, Caspase-3, and GADPH overnight at 4◦C. After washing in TBST, the membranes were incubated with their respective secondary antibodies for 1 hour.
Membranes were then incubated with SuperSignal West Femto Maximum Sensitivity Substrate (Pierce, Rockford, IL, USA) for 1 minute and imaged using a Gel Doc XR system (Bio-Rad). All of the antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
2.7. Statistics. All data are expressed as means of the standard deviation (SD). Statistical analysis was performed using the SPSS 13.0 software (SPSS, Chicago, IL, USA). Differences between the two groups were determined by paired-samples t-test or independent-samples t-test (two-tailed) as indicated. Differences among groups were analyzed by one-way analysis of variance (ANOVA). P < .05 was considered statistically significant.
3. Results
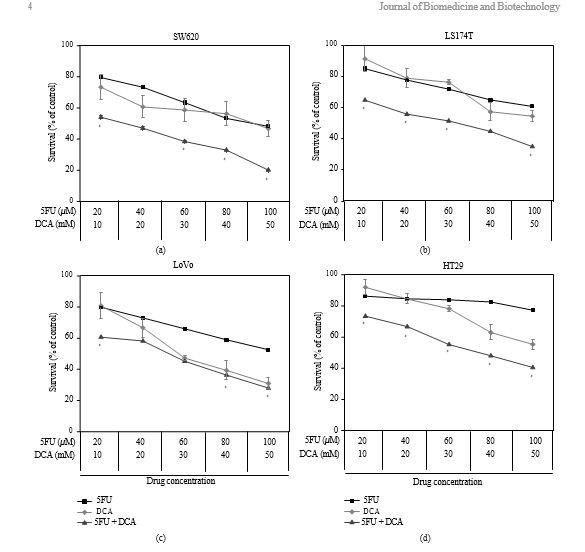
3.1. Viability of CRC Cells Treated with DCA Alone or in Combination with 5-FU. To determine the effect of DCA on CRC cells, cells were exposed to DCA (0–90 mM) for 48 hours. The result showed that inhibitory effect was dose-dependent. As shown in Figure 1, the inhibition of viability of cancer cell lines treated with 50 mM DCA was
as follows: SW620 (46.73% ± 5.21%), LoVo (30.94% ± 3.57%), LS174t (54.59% ± 3.93%), and HT29 (55.31% ± 3.35%). We treated cells with 5-FU (20–100 μM) and found that the viability of SW620, LoVo, LS174t was inhibited significantly except HT29, with its viability inhibition not obvious under 80 μM 5-FU. When treated with these two drugs simultaneously, the viability of the aforementioned CRC cells was significantly decreased compared with DCA or 5- FU alone (IC50 values are shown in Table 1).
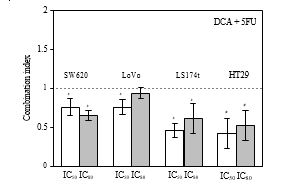
3.2. Synergistic Effects of DCA Combined with 5-FU in CRC Cell Lines. The viability of CRC cells were decreased in the presence of 5-FU and DCA. DCA potentiated the inhibitory effects of 5-FU on CRC cells, and the influence of DCA on effects of 5-FU was dose dependent (Figure 1). Interaction between 5-FU and DCA was analyzed with median effects method. The combination of DCA and 5-FU produced synergistic or additive effects depending on the range of cell kill level (Fa). All of four CRC cell lines indicated synergistic effect with 5-FU and DCA. The synergism was statistically significant in LS174t at inhibition levels of 50% and 80%, achieving CI values from 0.46–0.61, and in HT-29 cell line at inhibition levels of 50 and 80%, with CI values in the range from 0.42 to 0.52. In SW620 and LoVo cells, CI values at 50% and 80% inhibition levels were 0.64–0.75 and 0.75–0.94, respectively, (Figure 2).
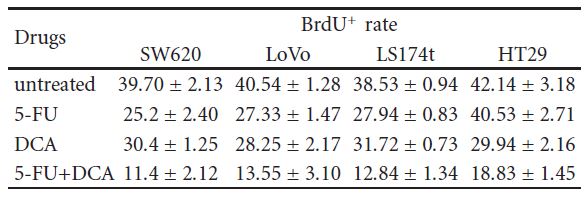
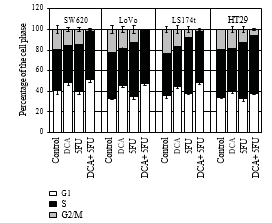
3.3. DCA Increased the Efficiency of Antiproliferative Effect of 5-FU. To confirm that decreased cell viability was due to reduced proliferation, immunocytochemistry, and flow cytometry were employed. Cells were treated with 10 mM DCA combined with 20 μM 5-FU. As expected, cells treated with DCA demonstrated reduced proliferation compared with untreated cells. The number of BrdU positive cells on four CRC cell lines after treatment with 5-FU and DCA were 11.4 ± 2.12, 13.55 ± 3.10, 12.84 ± 1.34, and 18.83 ± 1.45, respectively, which were lower than that treated with 5-FU or DCA alone (P < .01, see Table 2). In addition, treatment with DCA potentiated the cell cycle arrest in the G1 phase. When treated with DCA and 5-FU, cells blocked in the G1/S phase were more than that of incorporated with DCA or 5-FU alone (Figure 3).
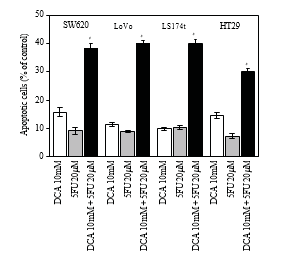
3.4. Increased Apoptosis Induced by DCA Combined with 5-FU in CRC Cells. To further investigate the decreased viability of CRC cells treated with a combination regimen, apoptosis was determined by flow cytometry. As shown in Figure 4, DCA alone increased the proportion of apoptosis CRC cells. When treated with 10 mM DCA, the apoptosis rates of four CRC cell lines were 15.72 ± 1.63%, 11.32 ± 0.74%, 9.77 ± 0.53%, and 14.52 ± 1.00%, respectively, while the apoptosis rates Table 1: IC50 values (concentrations required to reduce the viability of cells by 50% as compared with the control cells) were computed using linear or nonlinear regression (three parametric Hill func- tion) (R2> 0.9). They are presented as mean ± SD from at least three independent experiments. IC50 values of the studied drugs for inhibition of growth of various cell lines (cells were incubated with drugs for 48 hours).
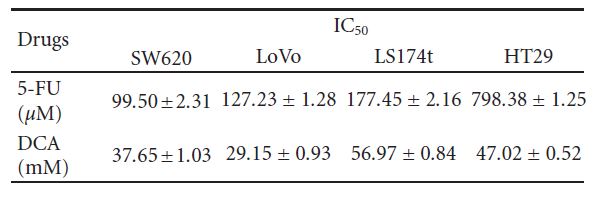
Table 2: SW620, LoVo, LS174t, HT29 cells, and 293T noncancerous controls were treated with 10 mM DCA and 20 μM 5-FU alone or in combination for 48 hours, and then pulsed with BrdU. Cells were then harvested and stained, and the numbers of BrdU+ cells were calculated as the mean number of the positive cells in eight different fields of view in one image (magnification, 400X). This calculation was repeated three times. ∗P < .05, compared with control. BrdU+ cells in different drug treatment (%, mean ± SD).
were 9.14 ± 119%, 8.82 ± 0.41%, 10.31 ± 0.71%, and 7.27± 0.96% with 5-FU. When combination of DCA and 5-FU was applied, the apoptosis rate was much higher than 5-FU or DCA alone (P < .05), which indicated apoptotic effect was increased via combination DCA and 5-FU (Figure 4).
3.5. Changes on Apoptosis-Associated Molecules Stimulated by DCA and 5-FU. To confirm that increased apoptosis induced by combination therapy was due to modified expressions of apoptosis-associated molecules, western blot assay was applied. In Figure 5, the results indicated that 5-FU or DCA decreased the expression of Bcl-2 in four CRC cell lines compared to PBS controls, and the combination of DCA and 5-FU decreased Bcl-2 expression significantly as compared with DCA or 5-FU alone. Conversely, the expressions of Bax and caspase-3 were significantly increased in the four CRC cell lines treated with combination of DCA and 5-FU compared to their single usage. The most obvious increasing of Bax expression was detected in LS174t cells, while in LoVo it appeared that caspase-3 expression increased most (Figure 5).
4. Discussion
In the present study, we demonstrate that DCA not only reduced cell viability and proliferation, but also have the synergistic antitumor efficiency with chemotherapeutic agent 5-FU in vitro in CRC cells.
Meanwhile, DCA has no significant effects on noncancerous cells. Furthermore, we showed that DCA-induced apoptosis contributes to its synergistic antitumor effect. Compared with DCA or 5- FU alone, the combination usage of these two drugs promotes apoptosis of CRC cells. 5-FU is a chemotherapy drug used to treat several types of cancer, including colorectal, breast, oesophagal, and stomach [15]. However, 5-FU–related toxicity is a serious and common issue for many cancer patients, with myelosuppression and gastrointestinal toxicity being the most commonly observed side effects [16]. The clinical activity of 5-FU is modest at standard doses, and in general, dosing is limited by the safety profile. As a result, we are usually left with the dilemma of making decisions about the therapeutic dose of 5-FU. Various strategies have been developed to enhance the clinical activity of 5-FU, such as biochemical modulation [17], alterations in the scheduling of administration [18], and the use of combination therapy [19–22]. DCA is an odourless, colourless, inexpensive, relatively nontoxic, small molecule. It has been in clinical use since 1969 for the treatment of lactic acidosis via boosting the ability of mitochondria to generate energy and reducing lactic acid accumulation [23].
DCA has been considered as a potential cancer therapy regimen since a Canadian group found that it caused regression in several cancers, including lung, breast, and brain tumours [24, 25]. When given to cancer cells, the cells switched from glycolysis to mitochondrial energy production. What’s more, functional mitochondria help cells recognize functional abnormalities and trigger cell death due to their inhibition of tumour growth and induction of apoptosis in certain cancer. In the present study, we found the same viability inhibitory effect in CRCs, which was dose-dependent and various with different degrees of tumour differentiation.
The results also indicated that low-dose DCA exerts a synergistic effect with chemotherapeutic agent 5- FU in halting growth of CRC cells, which was quantitatively analyzed according to Chou-Talalay method.
The results of cell proliferation also demonstrated that DCA enhanced the antiproliferational effects of 5-FU. The number of BrdU positive cells was decreased when treated with 5-FU or DCA, while the BrdU-stained cells were decreased significantly when treated with combination of 5- FU and DCA compared with their single usage. Meanwhile, the growth inhibition of CRC cell was accompanied by cell cycle arrest. Combination of DCA and 5-FU induced cell cycle arrest in G1/S phase in CRC cell lines, whereas 5-FU induced arrest in G1 phase was not obvious. The induction of cell cycle arrest may result from inhibitory ability to synthesize or repair DNA, which may lead to cell apoptosis.
DCA appeared to exert biochemical effects consistent with reversing the Warburg effect and killing cancer cells. We found that DCA-induced CRC cells apoptosis, which is orchestrated with the previous DCA studies. Importantly, the combination of DCA with 5-FU increased the number of apoptotic cells compared with 5-FU alone, demonstrating that DCA inhibited cell growth via apoptosis.
Many factors mediating apoptosis converge to activate the critical effector the caspase-3, which is considered as the key protease of the caspase family in mammalian cell apoptosis [26]. It always exists as a 23 kD inactive precursor in the cytoplasm, which is activated during apoptosis and takes part in apoptosis induced by multiple factors. Caspase- dependent apoptosis pathway mainly includes mitochondria pathway, death receptor pathway, and endoplasmic reticulum pathway [27].
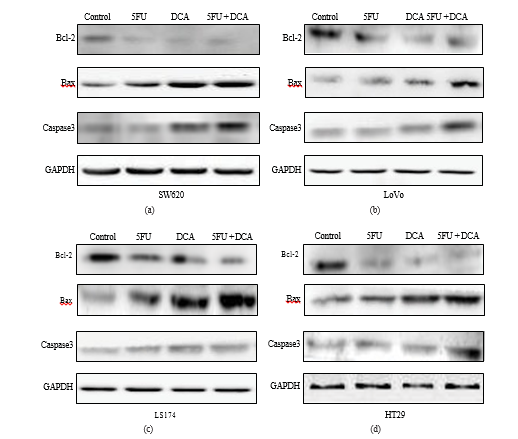
And the mitochondria pathway is controlled and regulated by the Bcl-2 family of proteins [28, 29], which are divided into two parts, the antiapoptotic members (Bcl-2) and proapoptotic members (Bax) [30]. Recent study indicates that the Bcl-2 inhibits apoptosis via inhibiting Bax removing to mitochondrial outer membrane [31]. We investigated the expression of caspase-3, Bcl-2, and Bax in protein level and the result of western blot showed that the expressions of caspase-3 and Bax were increased, while the expression of Bcl-2 was decreased in the combination of DCA with 5-FU treatment, comparing with the single treat- ments. These results suggested that the apoptosis induced by the combination of DCA and 5-FU might be related with caspase-dependent mitochondria pathway. Previous investi- gations suggested that the induction of apoptosis by DCA resulted from returning the dysfunction of the mitochondria and NFAT-Kv1.5 pathway [9, 12], which focused on the same point as present study that was mitochondria-mediated apoptosis.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (NSFC, no. 30873015). J. Tong and G. Xie contributed equally to this work.
References
[1] A. Jemal, R. Siegel, E. Ward, Y. Hao, J. Xu, and M. J. Thun, “Cancer statistics, 2009,” CA Cancer Journal for Clinicians, vol. 59, no. 4, pp. 225–249, 2009.[2] J. A. Meyerhardt and R. J. Mayer, “Drug therapy: systemic therapy for colorectal cancer,” The New England Journal of Medicine, vol. 352, no. 5, pp. 476–487, 2005.
[3] N. C. Tebbutt, E. Cattell, R. Midgley, D. Cunningham, and D. Kerr, “Systemic treatment of colorectal cancer,” European Journal of Cancer, vol. 38, no. 7, pp. 1000–1015, 2002.
[4] M. Gusella, A. C. Frigo, C. Bolzonella et al., “Predictors of survival and toxicity in patients on adjuvant therapy with 5- fluorouracil for colorectal cancer,” British Journal of Cancer, vol. 100, no. 10, pp. 1549–1557, 2009.
[5] D. Hanahan and R. A. Weinberg, “The hallmarks of cancer,” Cell, vol. 100, no. 1, pp. 57–70, 2000.
[6] Z. Chen,W. Lu, C. Garcia-Prieto, and P.Huang, “TheWarburg effect and its cancer therapeutic implications,” Journal of Bioenergetics and Biomembranes, vol. 39, no. 3, pp. 267–274, 2007.
[7] Y. Chen, R. Cairns, I. Papandreou, A. Koong, andN. C. Denko, “Oxygen consumption can regulate the growth of tumors, a new perspective on theWarburg effect,” PLoS ONE, vol. 4, no. 9, Article ID e7033, 2009.
[8] A. Aynsley Green, A. M. Weindling, G. Soltesz, and P. A. Jenkins, “Transient lactic acidosis and hyperalaninaemia associated with neonatal hyperinsulinaemic hypoglycaemia: the effects of dichloroacetate (DCA),” European Journal of Pediatrics, vol. 141, no. 2, pp. 114–117, 1983.
[9] J. Y. Y. Wong,G. S. Huggins,M. Debidda, N. C.Munshi, and I. De Vivo, “Dichloroacetate induces apoptosis in endometrial cancer cells,” Gynecologic Oncology, vol. 109, no. 3, pp. 394– 402, 2008.
[10] W. Cao, S. Yacoub, K. T. Shiverick et al., “Dichloroacetate (DCA) sensitizes both wild-type and over expressing bcl-2 prostate cancer cells in vitro to radiation,” Prostate, vol. 68, no. 11, pp. 1223–1231, 2008.
[11] E. D. Michelakis, L. Webster, and J. R. Mackey, “Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer,” British Journal of Cancer, vol. 99, no. 7, pp. 989–994, 2008.
[12] S. Bonnet, S. L. Archer, J. Allalunis-Turner et al., “A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth,” Cancer Cell, vol. 11, no. 1, pp. 37–51, 2007.
[13] T. C. Chou and P. Talalay, “Analysis of combined drug effects: a new look at a very old problem,” Trends in Pharmacological Sciences, vol. 4, pp. 450–454, 1983.
[14] T. C. Chou and P. Talalay, “Quantitative analysis of doseeffect relationships: the combined effects of multiple drugs or enzyme inhibitors,” Advances in Enzyme Regulation, vol. 22, pp. 27–55, 1984.
[15] D. B. Longley, D. P. Harkin, and P. G. Johnston, “5- Fluorouracil: mechanisms of action and clinical strategies,” Nature Reviews Cancer, vol. 3, no. 5, pp. 330–338, 2003.
[16] M. W. Saif, A. Choma, S. J. Salamone, and E. Chu, “Pharmacokinetically guided dose adjustment of 5-fluorouracil: a rational approach to improving therapeutic outcomes,” Journal of the National Cancer Institute, vol. 101, no. 22, pp. 1543–1552, 2009.
[17] C.G. Leichman, K. Chansky, J. S. Macdonaldet al., “Biochemical modulation of 5-fluorouacil through dihydropyrimidine dehydrogenase inhibition: a Southwest OncologyGroup phase II trial of eniluracil and 5-fluorouracil in advanced resistant colorectal cancer,” Investigational New Drugs, vol. 20, no. 4, pp. 419–424, 2002.
[18] F. A. Levi, R. Zidani, J. M. Vannetzel et al., “Chronomodulated versus fixed-infusion-rate delivery of ambulatory chemotherapy with oxaliplatin, fluorouracil, and folinic acid (leucovorin) in patients with colorectal cancer metastases: a randomized multi-institutional trial,” Journal of the National Cancer Institute, vol. 86, no. 21, pp. 1608–1617, 1994.
[19] G. Melen-Mucha, E. Balcerczak, S. Mucha, M. Panczyk, S. Lipa, and M. Mirowski, “Expression of p65 gene in experimental colon cancer under the influence of 5-fluorouracil given alone and in combination with hormonal modulation,” Neoplasma, vol. 51, no. 4, pp. 319–324, 2004.
[20] F. Richards II, L. D. Case, and D. R. White, “Combination chemotherapy (5-fluorouracil, methyl-CCNU, mitomycin C) versus 5-fluorouracil alone for advanced previously untreated colorectal carcinoma. A phase III study of the piedmont oncology association,” Journal of Clinical Oncology, vol. 4, no. 4, pp. 565–570, 1986.
[21] A. Aquino, S. P. Prete, J. W. Greiner et al., “Effect of the combined treatment with 5-fluorouracil, γ-interferon or folinic acid on carcinoembryonic antigen expression in colon cancer cells,” Clinical Cancer Research, vol. 4, no. 10, pp. 2473– 2481, 1998.
[22] S. Obi, H. Yoshida, R. Toune et al., “Combination therapy of intraarterial 5-fluorouracil and systemic interferon-alpha for advanced hepatocellular carcinoma with portal venous invasion,” Cancer, vol. 106, no. 9, pp. 1990–1997, 2006.
[23] P. W. Stacpoole, “Review of the pharmacologic and therapeutic effects of diisopropylammonium dichloroacetate (DIPA),” The Journal of Clinical Pharmacology, vol. 9, no. 5, pp. 282– 291, 1969.
[24] S. Bonnet, S. L. Archer, J. Allalunis-Turner et al., “A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth,” Cancer Cell, vol. 11, no. 1, pp. 37–51, 2007.
[25] E. D.Michelakis, G. Sutendra, P.Dromparis et al., “Metabolic modulation of glioblastoma with dichloroacetate,” Science Translational Medicine, vol. 2, no. 31, pp. 31–ra34, 2010.
[26] T. Fernandes-Alnemri, G. Litwack, and E. S. Alnemri, “CPP32, a novel human apoptotic protein with homology to Caenorhabditis elegans cell death protein Ced-3 and mammalian interleukin-1β-converting enzyme,” The Journal of Biological Chemistry, vol. 269, no. 49, pp. 30761–30764, 1994.
[27] H. Mehmet, “Caspases find a new place to hide,” Nature, vol. 403, no. 6765, pp. 29–30, 2000.
[28] E. Yang and S. J. Korsmeyer, “Molecular thanatopsis: a discourse on the BCL2 family and cell death,” Blood, vol. 88, no. 2, pp. 386–401, 1996.
[29] D. R. Green and J. C. Reed, “Mitochondria and apoptosis,” Science, vol. 281, no. 5381, pp. 1309–1312, 1998.
[30] J. C. Reed, “Double identity for proteins of the Bcl-2 family,” Nature, vol. 387, no. 6635, pp. 773–776, 1997.
[31] B. Antonsson, S. Montessuit, B. Sanchez, and J. C. Martinou, “Bax is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells,” The Journal of Biological Chemistry, vol. 276, no. 15, pp. 11615– 11623, 2001.