Jason Y.Y. Wong1, Gordon S. Huggins2, Marcella Debidda4, Nikhil C. Munshi4, and Immaculata De Vivo1,3
1 Channing Laboratory, Department of Medicine, Brigham and Women’s Hospital and Harvard Medical School, Boston Massachusetts.
2 Molecular Cardiology Research Institute, Tufts-New England Medical Center, Boston Massachusetts.
3 Program in Molecular and Genetic Epidemiology, Harvard School of Public Health, Boston Massachusetts.
4 The Jerome Lipper Multiple Myeloma Center, Department of Medical Oncology, Dana-Farber Cancer Institute, Harvard Medical School, Boston Massachusetts.
Correspondence: Immaculata De Vivo, Brigham and Women’s Hospital, Department of Medicine, Channing Laboratory, 181 Longwood Ave, Boston, MA, 02115, USA. Phone: 617-525-2094. Fax: 617-525-2008. [email protected].
Received: 14 September 2020
Accepted: 4 December 2020
Published: 9 December 2020
Abstract
Purpose: A recent landmark study demonstrated that Dichloroacetate (DCA) treatment promoted apoptosis in lung, breast, and glioblastoma cancer cell lines by shifting metabolism from aerobic glycolysis to glucose oxidation coupled with NFAT-Kv1.5 axis remodeling. The objective of this study was to determine whether DCA induces apoptosis in endometrial cancer cells and to assess apoptotic mechanism.
Methods: A panel of endometrial cancer cell lines with varying degrees of differentiation was treated with DCA and analyzed for apoptosis via flow cytometry. Biological correlates such as gene expression, intracellular Ca2+, and mitochondrial membrane potential were examined to assess apoptotic mechanism.
Results: Initiation of apoptosis was observed in five low to moderately invasive cancer cell lines including Ishikawa, RL95-2, KLE, AN3CA, and SKUT1B while treatment had no effect on non-cancerous 293T cells. Two highly invasive endometrial adenocarcinoma cell lines, HEC1A and HEC1B, were found to be resistant to DCA-induced apoptosis. Apoptotic responding cell lines had a significant increase in early and late apoptotis, a decrease in mitochondrial membrane potential, and decreased Survivin transcript abundance, which are consistent with a mitochondrial-regulated mechanism. DCA treatment decreased intracellular calcium levels in most apoptotic responding cell lines which suggests a contribution from the NFAT-Kv1.5-mediated pathway. DCA treatment increased p53 upregulated modulator of apoptosis (PUMA) transcripts in cell lines with an apoptotic response, suggesting involvement of a p53-PUMA-mediated mechanism.
Keywords: Dichloroacetate; Endometrial; Cancer; Apoptosis; Mitochondria
Conflict of Interest Statement: The authors declare there are no conflicts of interest.
Conclusions: Dichloroacetate effectively sensitizes most endometrial cancer cell lines to apoptosis via mitochondrial, NFAT-Kv1.5, and PUMA-mediated mechanisms. Further investigation of the cancer therapeutic potential of DCA is warranted.
MMP is further confirmed via TMRM fluorescent staining in a DCA dose-response experiment. Error bars represent standard error from 2 independent experiments performed in triplicate wells.
INTRODUCTION
Endometrial cancer (EC) is a neoplasia of the epithelial lining of the uterine corpus. It is the most common gynecologic malignancy in the United States and the fourth leading cause of cancer death in the country among women [1]. There are few therapeutic options without serious drawbacks for those with recurrent or metastatic endometrial cancer. Chemotherapy for metastatic disease has high rates of toxicity, neuralgia, and cardiac complications[2, 3]. The impetus in future cancer therapy development will be to reduce serious adverse effects while demonstrating comparable or improved efficacy to existing treatments.
Aerobic glycolysis, also known as the ‘Warburg Effect’, is a unique property of most cancers. This phenomenon is characterized by increased glucose uptake and reliance on glycolysis for ATP production despite an available oxygen source [5]. Aerobic glycolysis is believed to be a result of mitochondrial dysfunction which confers apoptotic resistance in cancer cells [6]. This apoptotic resistance is due to hyperpolarization of the mitochondrial membrane which prevents the release of pro-apoptotic mediators from the mitochondria to the cytoplasm [4]. Hyperpolarized mitochondrial membranes are characteristic of most carcinomas and its reversal is associated with initiation of apoptosis [7,8].
Therapeutic targeting of aerobic glycolysis is a novel means in which to target cancer cells. The key regulator of cellular metabolism is pyruvate dehydrogenase (PDH) which in turn is inhibited by pyruvate dehydrogenase kinase (PDK). A recent study showed that PDK activity in cancer cell lines can be down-regulated by DCA [4]. Metabolic targeting by DCA involves two synergistic mechanisms, the proximal and distal pathways [4]. In the proximal (mitochondrial-regulated) pathway, DCA binds to PDK and attenuates inhibition of PDH activity. The increased PDH activity shifts metabolism from glycolysis to glucose oxidation and decreases mitochondrial membrane potential (MMP) hyperpolarization, which opens mitochondrial transition pores (MTPs). This allows for the translocation of reactive oxygen species (ROS) and cytochrome c from the mitochondria to the cytoplasm, subsequently inducing apoptosis through the activation of caspases [4]. In the distal (NFAT-Kv1.5) pathway, translocated ROS dilates Kv1.5 potassium ion channels on the plasma membrane. The expulsion of potassium ions hyperpolarizes the cell, preventing voltage-dependent Ca2+ entry. The decreased intracellular Ca2+ level inhibits the activation of NFAT, which further increases Kv1.5 expression; creating a positive feedback loop ultimately resulting in reduced tonic inhibition of caspases [4].
There is substantial cross-talk between the mitochondrial and the p53-mediated apoptotic pathways. p53-upregulated modulator of apoptosis (PUMA) is one such pro-apoptotic protein that bridges the interaction between mitochondrial and p53 tumor suppressor-mediated mechanisms. PUMA is a member of the BH3-only family of proteins whose expression is transcriptionally regulated by p53 [9–11]. Upon activation by various apoptotic stimuli, PUMA translocates to the mitochondrial membrane where it antagonizes pro-survival Bcl-2 proteins by binding to its BH3 domain, inducing cytochrome c release, and promoting apoptosis [10,12]. In recent knock-out studies, PUMA has been determined to be a critical mediator of p53-dependent apoptosis in murine thymocytes and human colorectal cancer cells [13,14].
Dichloroacetate has been shown in numerous studies to promote glucose oxidation in various mitochondrial disorders [15,16]. Additionally, DCA treatment was found to have milder side effects in clinical studies of mitochondrial encephalomyopathies compared to those of current endometrial cancer therapies [17]. To date, the effect of DCA has been studied in a limited number of cancer cell lines and our understanding of alternative apoptotic mechanisms regulated by DCA is deficient. The purpose of our study was to determine whether DCA sensitizes a panel of endometrial cancer cell lines to apoptosis and to assess the contribution of the mitochondrial-regulated, NFAT-Kv1.5, and PUMA mechanisms in the apoptotic process by examining biological correlates.
Materials and Methods
Cell Culture
AN3CA, SKUT1B, RL95-2, KLE, HEC1A, and HEC1B cell lines were purchased from American Type Culture Collection (Manassas, VA) and the Ishikawa cell line was purchased from Sigma-Aldrich (St. Louis, MO). The 293T kidney epithelial cells that served as non-cancerous, healthy controls were provided by Nikhil Munshi. MCF7 breast epithelial adenocarcinoma was a gift of Ramon Parsons (Columbia University). Cell lines were propagated as per distributor’s specified conditions. Cell lines were maintained in a 37°C, 5% CO2 humidified incubator. DMEM, McCoy’s 5A, MEM, and DMEM-F12 growth media along with penicillin-streptomycin and insulin supplements were purchased from Gibco-Invitrogen (Carlsbad, CA). Dichloroacetate (Alfa Aesar, Ward Hill, MA) was dissolved to a 1M working solution, filter-sterilized, and subsequently diluted to treatment concentrations in growth media.
Cell Viability Assay
Cell viability was measured using CellTiter-Blue reagent (Promega) which measures the ability of healthy viable cells to metabolize a Resazurin substrate to a fluorescent Resorufin product. Briefly, 3×104 cells of each cell line were plated into opaque-walled 96-well tissue culture plates and incubated in standard growth conditions overnight to 60–70% confluence. The media in each well was then replaced with fresh growth media containing increasing concentrations of DCA (0 mM, 1 mM, 5 mM, 10 mM). Each well was performed in triplicate in two or more independent experiments for each cell line. Following treatment, the plates were incubated for 40 hours at 37°C after which 20 µL of Resazurin substrate was added directly to each well and incubated for an additional 3 hours. The plates were then read on a Molecular Devices Gemini XPS plate reader (Sunnyvale, CA) at 560/590 nm.
Apoptosis Assays
Flow cytometry with Annexin-V-FITC (BD Bioscience, San Jose, CA) and 7-amino-actinomycin D (7-AAD) staining was used to determine whether treatment specifically induces early apoptosis. Briefly, 5×105 cells for each cell line were seeded into 6-well tissue culture plates and incubated overnight to 60–70% confluence under standard growth conditions. Media for each cell line was then replaced with fresh growth media with and without a 10 mM dose of DCA. Treatment groups for each cell line were replicated three times. The cells were then incubated for 40 hours at 37°C and harvested with 0.25% Trypsin-EDTA (Invitrogen, Carlsbad, CA). Cells were washed with 1X PBS and subsequently stained as per manufacturer’s protocol. Flow cytometry was performed on a BD FACSCanto II (BD Bioscience) and data was analyzed on FlowJo 7.2.2 (Tree Star, Ashland, OR) and BD FACSDiva 6.0 software (BD Bioscience).
The Apoptag Peroxidase Terminal dUTP Nick-end Labeling (TUNEL) assay kit (Millipore, Billerica, MA) was used to visualize apoptotic cells that had undergone caspase-dependent genomic fragmentation. Briefly, 5×104 cells of several representative endometrial cancer cell lines were seeded and propagated on 4-well chamber slides (Nunc, Rochester, NY) overnight. The media in each well was then replaced with fresh growth media with or without 10 mM DCA. After 48 hours, the cells were fixed with 1% paraformaldehyde and stained as per manufacturer’s protocol. Staining was performed by the Dana Farber – Harvard Cancer Center Pathology Core facility. Images were captured at 40X objective on a Zeiss Axioskop 2 Plus microscope (Thornwood, NY) using AxioVs40 v.4.4.1.0 software at 24-bit RGB.
Cell Proliferation Assay
Flow cytometry using bromodeoxyuridine (BrdU) (BD Bioscience) and 7-AAD staining was employed to measure cell proliferation. Briefly, several representative endometrial cancer cell lines were propagated as outlined above for the Annexin-V assay. The cells were then serum-starved for 8 hours in growth media containing 0.5% FBS to reset the cell cycle to G0 phase. The media was subsequently changed to normal growth media with and without 10 mM DCA treatment. After 24 hours, the cells were pulsed for 2 hours with 10 µM BrdU in growth media. The cells were then harvested, stained, and analyzed as per manufacturer’s protocol.
Mitochondrial Membrane Potential Assays
Mitochondrial membrane potential was detected using the Mitocapture Apoptosis Detection Kit (Calbiochem). Growth, treatment, and experimental layout of the cell lines were identical to the Annexin-V assay described above. After a 24-hour incubation period following treatment with and without 10 mM DCA, the cells were harvested and washed with 1X PBS and stained with Mitocapture reagent as per manufacturer’s protocol and analyzed via flow cytometry. A modified version of a protocol involving staining mitochondria with tetramethyl rhodamine methyl ester (TMRM) (Invitrogen, Carlsbad, CA) described elsewhere, was also used to assess MMP [18]. Briefly, the cells were propagated and treated exactly as the cell viability assay described above. After a 24-hour incubation period, 5×104 cells were isolated, washed in 1X PBS, and resuspended in Hanks buffered salt solution (HBSS) (Sigma-Aldrich, St. Louis, MA) with 50 nM TMRM and incubated for 30 mins at 37°C. The cells were transferred to an opaque 96-well plate and fluorescence was measured at 530/620 nm at 37°C using a plate reader.
Intracellular Calcium Levels
Intracellular calcium levels were measured using the FLUO-4 NW Calcium Assay (Invitrogen). Briefly, 3×104 cells for each cell line were plated onto individual opaque-walled 96-well tissue culture plates and incubated in standard growth conditions for 8 hours. The media in each well was then replaced with fresh growth media containing increasing concentrations of DCA. Each treatment group was replicated in 4 wells in at least 2 independent experiments. Following 8-hour incubation, the cells in each well were treated with FLUO-4 reagent as per manufacturer’s protocol. The plates were then read on a fluorescent plate reader at 494/516 nm.
Real Time PCR
Real-time quantitative PCR was used to detect the abundance of endogenous Survivin and PUMA transcripts. A total of 1×106 cells for each cell line were seeded and grown in 10cm tissue culture plates overnight. The media was then replaced with fresh growth media with or without 10 mM DCA treatment. After incubating for 40 hours, 3×106 cells for each plate were harvested and total RNA was extracted using RNeasy Plus Mini Kit (Qiagen, Valencia, CA) as per manufacturer’s protocol. First-strand cDNA was synthesized with 1000 ng of total RNA and Oligo dT primers using Superscript III Reverse Transcriptase (Invitrogen) as per manufacturer’s protocol. The cDNA product was then treated with RNase H for 20 minutes at 37°C and diluted to100 ng/µL. Concentrations of RNA and cDNA were precisely determined using a Nanodrop ND-1000 spectrophotometer (Wilmington, DE).
The primer sequences for Survivin were forward 5’-AAGAACTGGCCCTTCTTGGA-3’ and reverse 5’- CAACCGGACGAATGCTTTT-3’ (Primerbank). The primers sequences for PUMA and the RPLP0 housekeeping gene were described in previous studies [19, 20]. The reaction mixtures consisted of 1X Applied Biosystems SYBR Green PCR mix (Foster City, CA), 1.5 mM MgCl2, 0.42 mM dNTPs, 5U ABI Amplitaq Gold, 200 ng of cDNA template, and 333 nM of forward and reverse primers. Reactions were performed in triplicate in two replicate experiments. The cycling conditions were 1 cycle at 95C for 10:00, 33x cycles of 95C for 0:30, 55C for 0:30, and 72C at 0:30. A five-point standard curve for the reactions had linear slopes of −3.2 +/− 0.1 with correlation coefficients (r2) above 0.985. The assay was performed with an ABI 7300 Real-Time PCR System (Foster City, CA). Relative quantification of the target transcripts normalized to RPLP0 was assessed with ABI 7300 Real-Time PCR Systems RQ Study Software using the comparative Ct method.
Statistical Analysis
Student’s t-test and One-way ANOVA were used to evaluate differences between treatment arms. A p-value below 0.05 was considered significant. Analysis was performed with Microsoft Excel 2007 (Redmond, WA). Graphs were created using GraphPad Prism 5 (San Diego, CA).
Results
DCA Reduces Endometrial Cancer Cell Viability in a Dose-Dependent Manner
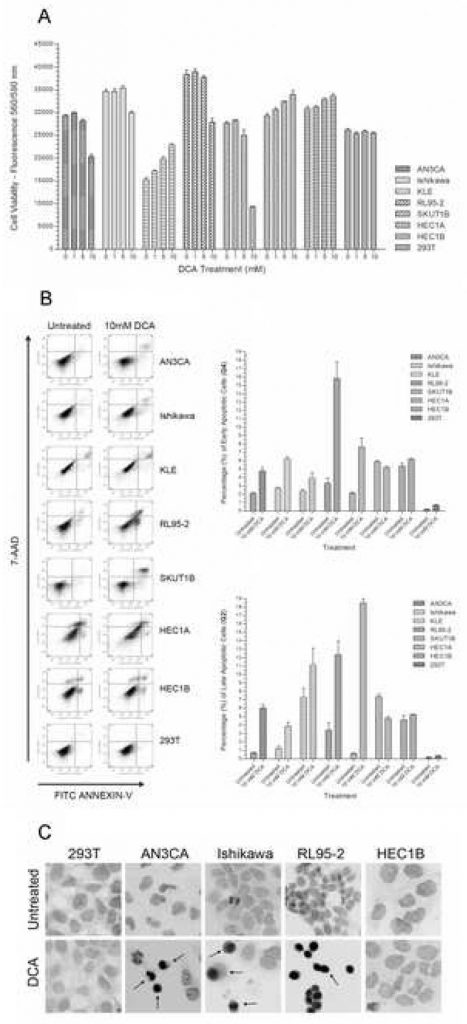
To determine the effect of DCA on the viability of endometrial cancer cells, each cell line was grown in culture with increasing doses of DCA. In a panel of seven endometrial cancer cells lines, AN3CA, Ishikawa, RL95-2, and SKUT1B had a 15% – 75% decrease in viability with increasing DCA concentration (Figure 1A). Reduction in viability for most of the responding cell lines reached significance at the 10 mM dose. A comparison between the untreated group and the 10 mM DCA-treated group had p-values < 0.01 for AN3CA, Ishikawa, RL95-2, and SKUT1B. A marginally significant decrease in viability was seen at the 5mM dose for AN3CA and RL95-2. Therefore, an approximate effective minimal dose of DCA for these endometrial cell lines under the treatment period was determined to be between 5mM and 10mM. This DCA dose concentration and treatment time is within the effective range of previously published in vitro experiments [21]. A statistically significant increase in viability was observed with HEC1A, HEC1B, and KLE with increasing DCA concentrations (p < 0.02). As expected, no statistically significant difference in viability was observed in the 293T epithelial cells at the given DCA dose range and treatment period (p = 0.27).
The Effect of DCA on Endometrial Cancer Cell Proliferation is Cell Line-Dependent
To determine whether the observed decrease in cell viability with DCA treatment was due to a cell proliferation effect, a BrdU / 7-AAD staining was performed on several representative cell lines and analyzed via flow cytometry. No significant difference in cell cycle dynamics or proliferation was observed in Ishikawa, HEC1B, and 293T epithelial cells with DCA treatment (Table 1). In AN3CA, treatment with DCA increased proliferation, as indicated by significant increases in the number of cells in S and G2/M phases and fewer cells in G0/G1. In RL95-2 cells, treatment with DCA significantly decreased the proportion of cells in S and G2/M phases and increased cells in G0/G1 phase; indicating decreased proliferation and cell cycle arrest in either a senescent or quiescent state.
| Cell Line / Treatment | % S Phase | Stdev S Phase | % G0 / G1 Phase | Stdev G0/G1 | % G2 / M Phase | Stdev G2 / M Phase |
|---|---|---|---|---|---|---|
| 293T Untreated | 60.6 | 0.6 | 33.2 | 1.7 | 6.2 | 1.25 |
| 293T DCA | 58.1 | 2.2 | 31.7 | 2.2 | 10.2 | 0.34 |
| p-value | 0.15 | 0.14 | 0.03 | |||
| Ishikawa Untreated | 61.3 | 0.4 | 27.4 | 1.8 | 11.3 | 0.96 |
| Ishikawa DCA | 65.9 | 4.5 | 22.9 | 4.9 | 11.2 | 0.68 |
| p-value | 0.21 | 0.25 | 0.96 | |||
| HEC1B Untreated | 37.4 | 2.7 | 41.5 | 0.8 | 21.2 | 1.93 |
| HEC1B DCA | 42.4 | 1.5 | 34.8 | 3.6 | 22.8 | 2.15 |
| p-value | 0.19 | 0.08 | 0.47 | |||
| AN3CA Untreated | 43.5 | 0.6 | 50.6 | 2.0 | 5.9 | 1.80 |
DCA Promotes Apoptosis in Endometrial Cancer Cells
In order to determine whether the reduction of cell viability from DCA treatment was due to apoptosis rather than necrosis, an Annexin-V-FITC and 7-AAD cell staining was performed and analyzed using flow cytometry. Significant increases of 50% to 325% were observed in early apoptotic cells in AN3CA, Ishikawa, KLE, RL95-2 and SKUT1B (Figure 1B). Significant increases were also observed in late apoptotic cells of these cell lines (Figure 1B). RL95-2 had the greatest increase in early apoptotic cells, while KLE had the least significant increase. The increased percentage of late apoptotic cells in KLE was not statistically significant. There was no observed difference in the percentage of early and late apoptotic cells with treatment in HEC1B cells, while HEC1A cells had a slightly significant decrease in apoptotic cells. The 293T cell line did not undergo apoptosis with DCA treatment and the slight increase in the percentage of apoptotic cells was not significant (p=0.08).
Since the Annexin-V assay is primarily used to detect early apoptosis, a TUNEL assay was performed on several representative endometrial cancer cell lines to qualitatively confirm progression to late apoptosis by visualizing caspase-dependent DNA fragmentation. In concordance with the quantitative Annexin-V assay, increases in TUNEL-positive apoptotic cells were observed in AN3CA, Ishikawa, and RL95-2 (Figure 1C). No visual difference in TUNEL-positive cells was observed in HEC1B and 293T cells with 10 mM DCA treatment.
In order to determine if growth rate affects sensitivity of the endometrial cancer cell lines to DCA treatment, one apoptotic responder and non-responder, (Ishikawa and HEC1A respectively), were grown in serum-deprived conditions with 0.5% FBS, which resets cells to G0 phase and hinders proliferation. Serum starvation did not affect the proportion of early apoptotic cells in the Ishikawa cells with treatment compared to normal growth conditions. The percentage of early apoptotic cells increased from 3.17% +/− 0.21% SD in untreated to 6.20% +/− 1.04% SD p=0.05 in treated Ishikawa cells which was similar to results under normal growth conditions. The percentage of late apoptotic Ishikawa cells increased from 1.07% +/− 0.15% SD in untreated to 3.57% +/− 0.49% SD p=0.02 in treated cells. The HEC1A cell line showed no significant difference in early and late apoptotic cells. The percentage of HEC1A early apoptotic cells were 3.73% +/− 0.51% SD for untreated and 1.93% +/− 0.60 SD p=0.07 for treated cells. The percentage of HEC1A late apoptotic cells was 3.60% +/− 0.69% SD for untreated and 4.90% +/− 1.37 SD p=0.25 for treated.
Apoptosis is Mediated by Decreased Intracellular Calcium Levels
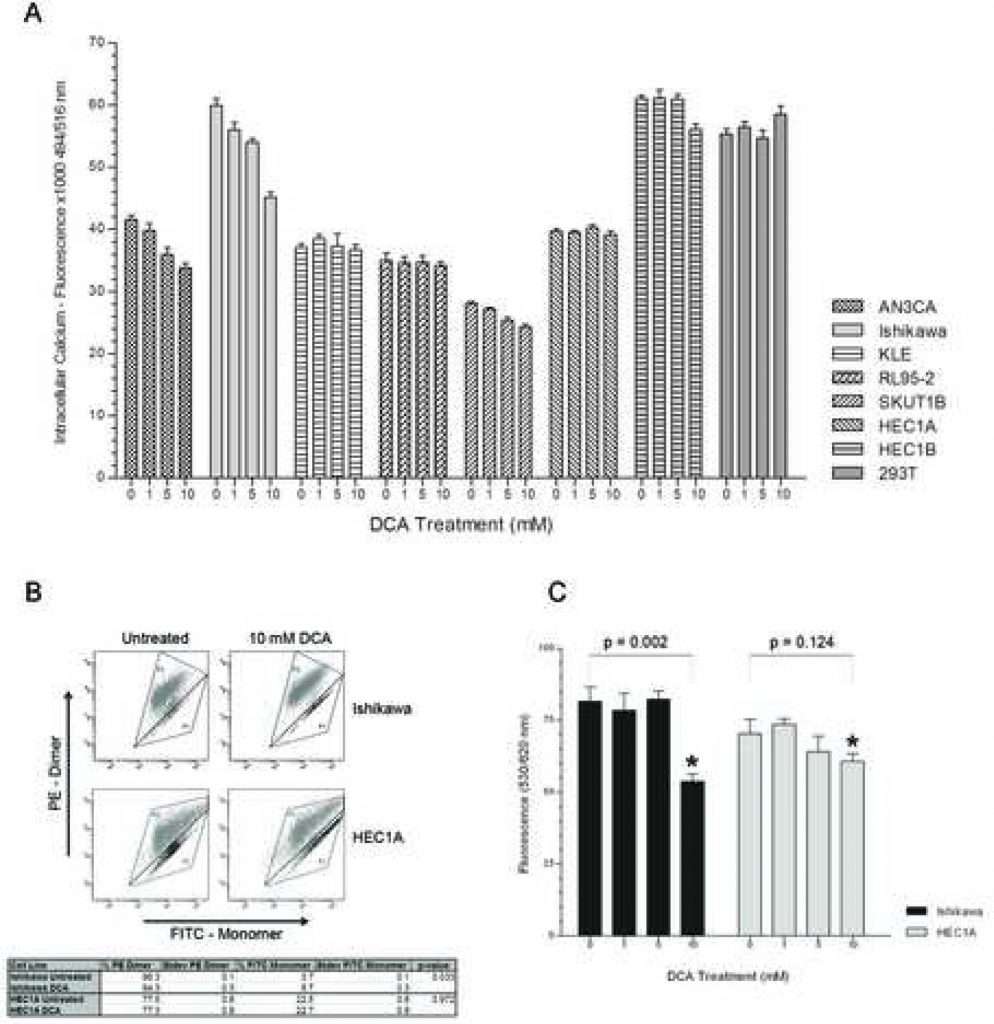
In order to determine whether the distal NFAT-Kv1.5 pathway contributes to DCA promotion of apoptosis, a dose-response experiment was performed to assess intracellular Ca2+ levels. AN3CA, Ishikawa, and SKUTB had decreasing intracellular calcium levels with increasing DCA doses (Figure 2A). The decreased intracellular calcium in these cell lines reached a significant level at a concentration of 5mM DCA with p < 0.03 for AN3CA, Ishikawa and SKUT1B. The KLE cell line, which showed the mildest apoptotic response, had an insignificant decrease in calcium levels with increasing DCA concentration. Interestingly, no difference in intracellular calcium levels between doses was detected with RL95-2, which had the greatest apoptotic response. HEC1A, which previously showed no apoptotic response, indeed had no difference in intracellular calcium levels at any treatment concentration. HEC1B also had no difference in calcium levels between 0, 1, and 5 mM DCA treatment groups and only a slight decrease is observed with the 10 mM treatment. There was no difference in intracellular calcium levels in 293T with increasing DCA concentration.
DCA Treatment Reduces Mitochondrial Membrane Hyper-polarization in Endometrial Cancer Cells that Undergo Apoptosis
To assess if DCA contributed to the initiation of apoptosis via a mitochondrial-regulated mechanism, the MMP of one apoptotic responder and non-responder, (Ishikawa and HEC1A respectively), were measured with and without treatment using FACS analysis. MitoCapture reagent is a cationic dye that depending on the extent of the mitochondrial transmembrane electrical potential, accumulates as green-emitting monomer in the cytoplasm or as a red-emitting dimer in hyperpolarized mitochondria of cancer cells [22, 23]. DCA treatment of the Ishikawa cell line reduced the percentage of 575 nm red-stained cells and increased the proportion of 525 nm green-stained cells which corresponds with its apoptotic response to treatment (Figure 2B). DCA treatment of the HEC1A cell line did not affect the percentage of red- and green-stained cells. Additionally, untreated HEC1A cells had a lower proportion of red-stained cells with hyper-polarized mitochondrial membranes compared to Ishikawa (77.5% +/− 0.6% SD vs. 96.3% +/− 0.1% SD, p < 0.01). There was no difference in MMP of the 293T non-cancerous control with treatment (data not shown). DCA modulation of MMP was further confirmed using TMRM staining in a dose-response experiment. The MMP of Ishikawa cells significantly decreased at the 10 mM DCA dosage (Figure 2C). There was no significant difference in the MMP of HEC1A cells at any treatment concentration.
DCA Decreases Survivin Expression
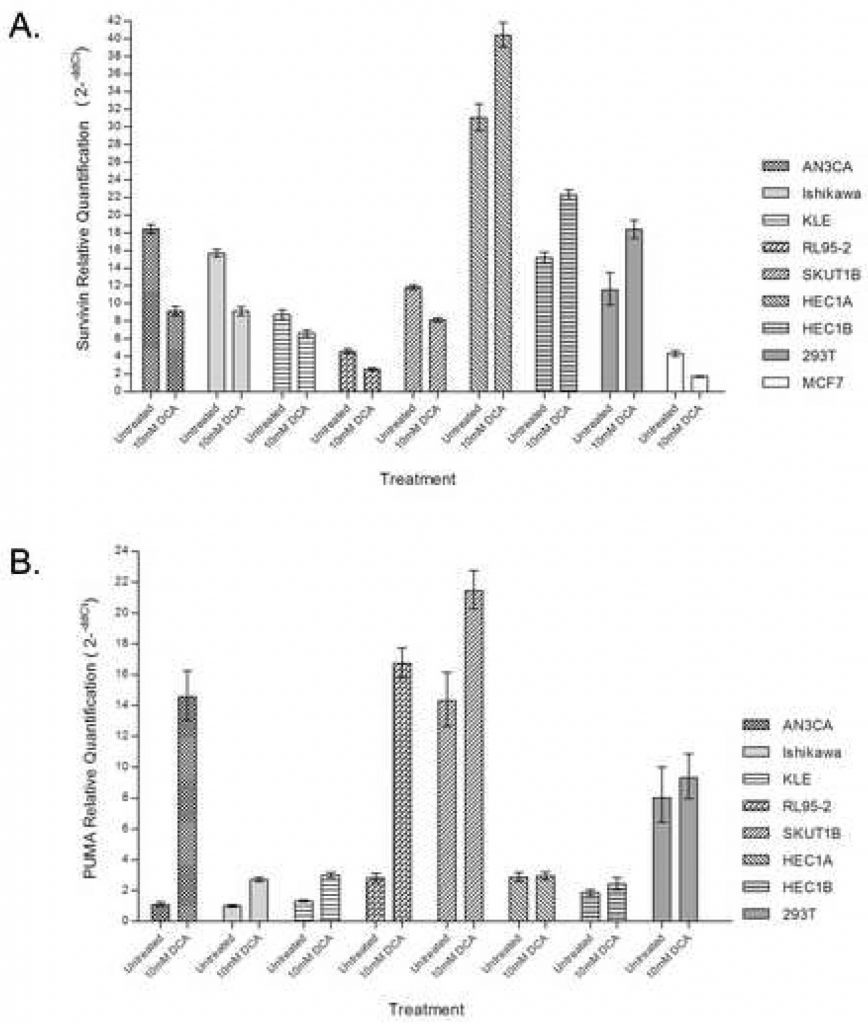
In order to further confirm the contribution of the proximal mitochondrial-regulated pathway to apoptotic response, real-time quantitative PCR was used to assess Survivin expression with and without DCA administration. Treatment of endometrial cancer cell lines with DCA resulted in a 25% – 50% decrease in the abundance of Survivin mRNA in cells that had an apoptotic response including AN3CA, Ishikawa, KLE, RL95-2, and SKUT1B (Figure 3A). RL95-2, which demonstrated the greatest increase in early apoptotic cells with treatment, also had the lowest endogenous Survivin transcript abundance. HEC1A and HEC1B, which previously had no apoptotic response to DCA, had 20 – 30% increases in Survivin transcripts with treatment. HEC1A had the greatest endogenous Survivin transcript abundance. The 293T control also showed increased transcript abundance with DCA treatment.
DCA Increases PUMA Expression
To determine the contribution of the PUMA pathway in DCA-induced apoptosis, real-time quantitative PCR was used to assess PUMA transcript abundance with and without DCA administration. Treatment of endometrial cancer cell lines with DCA dramatically increased the abundance of PUMA mRNA in cell lines that had an apoptotic response including AN3CA, Ishikawa, KLE, RL95-2, and SKUT1B (Figure 3B). The greatest degree of PUMA induction was observed in AN3CA and RL95-2 cells, which had 14-fold and 6-fold increases respectively. HEC1A and HEC1B, which previously had no apoptotic response to DCA, showed no difference in the quantity of PUMA transcript with treatment. No difference in the quantity of PUMA transcript was observed in 293T cells with treatment.
Discussion
In this study, we show that endometrial cancer cell death induced by DCA is regulated by two principal mechanisms; the mitochondrial-regulated and NFAT-Kv1.5 pathways. Furthermore, we demonstrated that DCA reduces endometrial cancer cell viability in a dose-dependent manner through the promotion of apoptosis while having no effect on non-cancerous 293T cells. Finally, we show that DCA treatment influences endometrial cancer cell survival through multiple molecular mechanisms, including regulation of mitochondrial membrane potential, intracellular Ca2+ levels, loss of Survivin expression, and PUMA induction.
The pro-apoptotic response of AN3CA, Ishikawa, and SKUT1B to DCA correlated with a dose-dependent decrease in intracellular Ca2+ levels, indicating the involvement of the NFAT-Kv1.5 mechanism. By comparison, RL95-2 (which had the greatest apoptotic response to DCA) and KLE (which had the mildest apoptotic response) did not show a difference in intracellular Ca2+ levels at any treatment concentration suggesting that the NFAT-Kv1.5 mechanism pathway is not involved in the apoptotic mechanism of these cell lines. Instead, DCA treatment arrested RL95-2 cells in G0/G1 phase of the cell cycle, a hallmark of p53 activation, strongly induced RL95-2 PUMA expression, and lowered expression of Survivin, a protein that plays a critical role in cell cycle regulation. [29].
Survivin is a transcriptionally regulated inhibitor of apoptosis that in response to disrupted MMP is discharged from the mitochondria to the cytoplasm, where it prevents caspase activation, inhibits apoptosis, and promotes tumor progression [26, 27]. Previous studies have shown a positive correlation between increased Survivin expression and endometrial carcinoma tumor grade [27,28]. We found that Survivin transcript abundance significantly decreased in all endometrial cancer cell lines that had an apoptotic response to DCA. Our results indicate that the mitochondrial-regulated pathway contributes to apoptotic response in DCA-sensitized endometrial cancer cell lines.
The PUMA transcript was significantly increased in all endometrial cancer cell lines that had an apoptotic response to DCA. The result may be indicative of contribution of the p53-PUMA pathway with the mitochondrial and ion channel mechanisms in DCA-induced apoptosis. The increased PUMA expression would further counteract the pro-survival effect of Bcl-2 on the mitochondrial membrane in the responding cell lines, allowing for increased translocation of apoptotic mediators from the mitochondria to the cytoplasm, thus promoting greater caspase activation and apoptosis.
Two cell lines, HEC1A and HEC1B, both highly invasive with increased drug resistance and higher tumor grade [24] compared to the other endometrial cell lines, were resistant to DCA. Indeed both cell lines showed increased Survivin expression, PUMA expression that was unaffected by DCA treatment, and a lower proportion of cells with hyperpolarized mitochondrial membranes. These findings suggest a lower reliance on aerobic glycolysis. We considered whether mutations in the DCA binding domain of the human PDK2 protein may explain differences in treatment response with HEC1A and HEC1B. However, no mutations were found in the 2 exons encoding the putative DCA binding domain by sequence analysis of all the cell lines (data not shown).
In summary, our study demonstrates that dichloroacetate is effective in sensitizing most low-to-moderately invasive endometrial cancer cells to apoptosis. Our collective data suggest that apoptosis is consistent with mitochondrial and NFAT-Kv1.5-mediated pathways. Additionally, our data suggest that the PUMA pathway may be involved in apoptotic promotion by DCA. Future investigations should focus on examining a broader range of cancer cell types in addition to determining the mechanisms that confer apoptotic resistance to DCA. The most potentially informative avenue to consider is functionally characterizing the role of the PUMA pathway in DCA-induced apoptosis.
Supplementary Material
| Endometrial Cancer Cell Line | Tumor Type | Degree of Differentiation | Invasiveness | p53 status |
| AN3CA | Epithelial Adenocarcinoma | Undifferentiated (30, 31) | Moderate (25, 32) | Mutant (33) |
| KLE | Adenocarcinoma | Poorly Differentiated (31, 34, 35) | Moderate (36) | Mutant (33) |
| Ishikawa | Epithelial Adenocarcinoma | Well Differentiated (25, 37) | Low (25) | Mutant (33) |
| RL95-2 | Epithelial Carcinoma | Moderately Differentiated (31, 35, 38) | Passage Number Dependent (39) | Mutant (codon deletion) (33) |
| SKUT1B | Mesodermal Leiomyosarcoma | Well Differentiated | Moderate-High (40) | n/a |
| HEC1A | Epithelial Adenocarcinoma | Moderately Differentiated (31, 41) | High (25, 42) | Mutant (33) |
| HEC1B | Epithelial Adenocarcinoma | Moderately Differentiated (31, 41) | High (25, 42) | Mutant (33) |
Acknowledgements
We would like to thank Dr. John Daley of the Dana Farber HemNeo Flow Cytometry core facility for technical training and Dr. Sabina Signoretti of the Dana Farber – Harvard Cancer Center Pathology Core facility for immunohistochemical support.
Grant Support: This project was supported by National Institutes of Health grant numbers CA082838 and CA101501.
REFERENCES
1 Rose P. Endometrial Carcinoma. New England Journal of Medicine. 1996;335(9):640–649. [PubMed] br>2 Ingram SS, Rosenman J, Heath R, Morgan TM, Moore D, Varia M. The predictive value of progesterone receptor levels in endometrial cancer. Int J Radiat Oncol Biol Phys. 1989;17(1):21–27. [PubMed] br>3 Randall ME, Filiaci VL, Muss H, et al. Randomized phase III trial of whole-abdominal irradiation versus doxorubicin and cisplatin chemotherapy in advanced endometrial carcinoma: a Gynecologic Oncology Group Study. J Clin Oncol. 2006;24(1):36–44. [PubMed] br>4 Bonnet S, Archer SL, Allalunis-Turner J, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11(1):37–51. [PubMed] br>5 Chen Z, Lu W, Garcia-Prieto C, Huang P. The Warburg effect and its cancer therapeutic implications. J Bioenerg Biomembr. 2007 [PubMed] br>6 Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006;66(18):8927–8930. [PubMed] br>7 Chen LB. Mitochondrial membrane potential in living cells. Annu Rev Cell Biol. 1988;4:155–181. [PubMed] br>8 Heerdt BG, Houston MA, Augenlicht LH. The intrinsic mitochondrial membrane potential of colonic carcinoma cells is linked to the probability of tumor progression. Cancer Res. 2005;65(21):9861–9867. [PubMed] br>9 Han J, Flemington C, Houghton AB, et al. Expression of bbc3, a pro-apoptotic BH3-only gene, is regulated by diverse cell death and survival signals. Proc Natl Acad Sci U S A. 2001;98(20):11318–11323. [PMC free article] [PubMed] br>10 Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7(3):683–694. [PubMed] br>11 Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell. 2001;7(3):673–682. [PubMed] br>12 Willis SN, Adams JM. Life in the balance: how BH3-only proteins induce apoptosis. Curr Opin Cell Biol. 2005;17(6):617–625. [PMC free article] [PubMed] br>13 Jeffers JR, Parganas E, Lee Y, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4(4):321–328. [PubMed] br>14 Yu J, Wang Z, Kinzler KW, Vogelstein B, Zhang L. PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc Natl Acad Sci U S A. 2003;100(4):1931–1936. [PMC free article] [PubMed] br>15 Abemayor E, Kovachich GB, Haugaard N. Effects of dichloroacetate on brain pyruvate dehydrogenase. J Neurochem. 1984;42(1):38–42. [PubMed] br>16 Lopaschuk GD, Saddik M. The relative contribution of glucose and fatty acids to ATP production in hearts reperfused following ischemia. Mol Cell Biochem. 1992;116(1–2):111–116. [PubMed] br>17 Stacpoole PW. The pharmacology of dichloroacetate. Metabolism. 1989;38(11):1124–1144. [PubMed] br>18 Wong A, Cortopassi GA. High-throughput measurement of mitochondrial membrane potential in a neural cell line using a fluorescence plate reader. Biochem Biophys Res Commun. 2002;298(5):750–754. [PubMed] br>19 Yan J, Jiang J, Lim CA, Wu Q, Ng HH, Chin KC. BLIMP1 regulates cell growth through repression of p53 transcription. Proc Natl Acad Sci U S A. 2007;104(6):1841–1846. [PMC free article] [PubMed] br>20 Bieche I, Parfait B, Tozlu S, Lidereau R, Vidaud M. Quantitation of androgen receptor gene expression in sporadic breast tumors by real-time RT-PCR: evidence that MYC is an AR-regulated gene. Carcinogenesis. 2001;22(9):1521–1526. [PubMed] br>21 Hassoun EA, Ray S. The induction of oxidative stress and cellular death by the drinking water disinfection by-products, dichloroacetate and trichloroacetate in J774.A1 cells. Comparative biochemistry and physiology Toxicology & pharmacology. 2003;135(2):119–128. [PubMed] br>22 Di Lisa F, Silverman HS, Hansford RG. Mitochondrial function and cell injury in single cardiac myocytes exposed to anoxia and reoxygenation. Transplant Proc. 1995;27(5):2829–2830. [PubMed] br>23 Piccoli C, Scrima R, Boffoli D, Capitanio N. Control by cytochrome c oxidase of the cellular oxidative phosphorylation system depends on the mitochondrial energy state. Biochem J. 2006;396(3):573–583. [PMC free article] [PubMed] br>24 Liang Y, O’Driscoll L, McDonnell S, et al. Enhanced in vitro invasiveness and drug resistance with altered gene expression patterns in a human lung carcinoma cell line after pulse selection with anticancer drugs. Int J Cancer. 2004;111(4):484–493. [PubMed] br>25 Sillem M, Prifti S, Koumouridis A, et al. Invasiveness corresponds to differentiation rather than to proteinase secretion in endometrial cancer cell lines. European Journal of Gynaecologic Oncology. 1999;20(5–6):367–370. [PubMed] br>26 Dohi T, Okada K, Xia F, et al. An IAP-IAP complex inhibits apoptosis. J Biol Chem. 2004;279(33):34087–34090. [PubMed] br>27 Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3(8):917–921. [PubMed] br>28 Erkanli S, Bolat F, Kayaselcuk F, Demirhan B, Kuscu E. COX-2 and survivin are overexpressed and positively correlated in endometrial carcinoma. Gynecol Oncol. 2007;104(2):320–325. [PubMed] br>29 Li F, Ambrosini G, Chu EY, et al. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 1998;396(6711):580–584. [PubMed] br>30 Rice LW, Stone RL, Xu M, et al. Biologic targets for therapeutic intervention in endometrioid endometrial adenocarcinoma and malignant mixed mullerian tumors. Am J Obstet Gynecol. 2006;194(4):1119–1126. discussion 26–28. [PubMed] br>31 Nagamani M, Stuart CA. Specific binding and growth-promoting activity of insulin in endometrial cancer cells in culture. Am J Obstet Gynecol. 1998;179(1):6–12. [PubMed] br>32 Zhao Y, Yamashita T, Ishikawa M. Regulation of tumor invasion by HOXB13 gene overexpressed in human endometrial cancer. Oncol Rep. 2005;13(4):721–726. [PubMed] br>33 Yaginuma Y, Westphal H. Analysis of the p53 gene in human uterine carcinoma cell lines. Cancer Res. 1991;51(24):6506–6509. [PubMed] br>34 Richardson GS, Dickersin GR, Atkins L, et al. KLE: a cell line with defective estrogen receptor derived from undifferentiated endometrial cancer. Gynecol Oncol. 1984;17(2):213–230. [PubMed] br>35 Carter CA, Parham GP. State of differentiation affects the response of endometrial adenocarcinoma cells to retinoic acid. Anticancer Research. 1997;17(3C):1973–1983. [PubMed] br>36 Yabushita H, Narumiya H, Hiratake K, et al. The association of transforming growth factor-beta 1 with myometrial invasion of endometrial carcinomas through effects on matrix metalloproteinase. J Obstet Gynaecol Res. 2000;26(3):163–170. [PubMed] br>37 Holinka CF, Hata H, Kuramoto H, et al. Responses to estradiol in a human endometrial adenocarcinoma cell line (Ishikawa) Journal of Steroid Biochemistry. 1986;24(1):85–89. [PubMed] br>38 Way DL, Grosso DS, Davis JR, et al. Characterization of a new human endometrial carcinoma (RL95-2) established in tissue culture. In Vitro. 1983;19(3 part 1):147–158. [PubMed] br>39 Sundareshan P, Hendrix MJ. Growth, morphologic, and invasive characteristics of early and late passages of a human endometrial carcinoma cell line (RL95-2) In Vitro Cell Dev Biol. 1992;28A(7–8):544–552. [PubMed] br>40 Colombatti A, Russo P, Cervi M, et al. Differential Expression of IRS-1 and IRS-2 in Uterine Leiomyosarcomas with Distinct Oncogenic Phenotypes: Lack of Correlation with Downstream Signaling Events. Sarcoma. 2002;6(3):89–96. br>41 Kuramoto H, Tamura S, Notake Y. Establishment of a cell line of human endometrial adenocarcinoma in vitro. Am J Obstet Gynecol. 1972;114(8):1012–1019. br>42 Sieuwerts AM, Klijn JG, Foekens JA. Assessment of the invasive potential of human gynecological tumor cell lines with the in vitro Boyden chamber assay: influences of the ability of cells to migrate through the filter membrane. Clin Exp Metastasis. 1997;15(1):53–62.
Related content: