Mao‑fa Zheng1, Si‑yu Shen, Wei‑da Huang
1Department of Biochemistry, School of Life Science, Fudan University, Handan Road 220, Shanghai, 200433, China.
Email: e-mail: [email protected]
Received: 16 May 2013
Accepted: 27 August 2013
Published: 17 September 2013
Abstract
Purpose: Capecitabine is one of the few chemotherapy drugs with high oral availability. Recently, sodium dichloroacetate (DCA) has shown great potential as an anticancer agent. In the present study, we assessed the anticancer effect of DCA in combination with capecitabine for cancers that modestly expressed TP.
Methods: A mouse B16 melanoma allograft and a human non-small cell lung cancer A549 xenograft were used to assess the effect of DCA and capecitabine combined treatment. Histology and immunohistochemistry were used to detect the apoptosis and proliferation of cancer cells. Real-time PCR and Western blot were carried out to detect the expression of TP and caspases, respectively.
Results: For the first time, we report that DCA increased the antitumor effects of capecitabine in a mouse B16 allograft and a human A549 xenograft by promoting apoptosis of tumor cells. DCA has little effect on the expression of TP.
Conclusions: Our finding suggests that DCA in combination with capecitabine might be potential as a new therapeutic regimen against some cancers.
Keywords: DCA, Capecitabine, Combination, Antitumor effect
© Springer-Verlag Berlin Heidelberg 2013
Cancer Chemother Pharmacol (2013) 72:1031–1041
DOI: 10.1007/s00280-013-2281-z
INTRODUCTION
Sodium dichloroacetate (DCA) is a small molecular salt of dichloroacetic acid with a molecular weight of 150 Da. DCA inhibits the activity of pyruvate dehydrogenase kinase, thus activating the mitochondrial enzyme complex pyruvate dehydrogenase [1] and converting the glycolytic metabolic pathway to oxidative phosphorylation. In the past 40 years, DCA has been used as an orphan drug in the treatment for children congenital lactic acidosis and other lactic acidosis complicated by other diseases [2] and has shown high efficacy and low toxicity in both preclinical and clinical trials [3]. Recently, DCA has shown great potential as an anticancer agent because of the similarities in metabolic remodeling of some tumor cells to that occurring during lactic acidosis [4]. Cancer cells, especially cancer stem cells (CSCs), resist apoptosis by producing energy through glycolysis and lactic acid fermentation, rather than oxidative phosphorylation, because of the hypoxic nature of the tumor microenvironment, a phenomenon known as the Warburg effect [5,6]. After oral administration, DCA has been shown to restore mitochondrial function and selectively promote tumor cell apoptosis in a mitochondrial-dependent pathway [7,8]. The therapeutic activities of DCA against glioblastoma have been tested in clinical trials (NCT00540176) and shown some positive results [9]. But phase II trial NCT01029925 to determine the response rate of oral dichloroacetate in patients with recurrent and/or metastatic and pretreated breast and non-small cell lung cancer was terminated due to higher than expected risk and safety concerns. So, the clinical utility of DCA for cancer control needs more careful estimation.
As an apoptosis sensitizer, DCA has also been used in combination with other cancer therapies. Cao et al. [10] reported that DCA sensitized prostate cancer cells to radiation in vitro. Xiao et al. [11] determined that DCA enhanced tumor cell death when combined with an oncolytic adenovirus expressing the tumor suppressor MDA-7/IL-24. Recently, metabolic-targeting therapy with DCA has been demonstrated as a novel treatment strategy to improve the outcome of photodynamic therapy [12]. Tong et al. [13] found that DCA and 5-fluorouracil showed synergistic antitumor effect in colorectal cancer cells in vitro. However, there are still controversial results and doubts regarding the use of DCA alone or in combination with other drugs. Shahrzad et al. [14] showed that DCA reduced apoptosis of cancer cells under hypoxic conditions both in vitro and in vivo. Heshe et al. [15] warned that DCA reduced the cytotoxicity of some standard anticancer drugs, such as cisplatin and doxorubicin, but did not affect the activity of temozolomide in 7 of 10 cell lines in their study. These conflicting results imply that the use of DCA alone or in combination with other therapies may be cancer-type-specific and agent-specific.
Capecitabine is one of the few chemotherapy drugs with high oral availability and is licensed as a first-line treatment for metastatic rectal cancer or as an alternative treatment for metastatic breast cancer when combined with docetaxel [16,17]. Capecitabine is a prodrug of 5-fluorouracil (5-FU) and requires 3 enzymatic reactions for a final conversion to 5-FU in tumor cells. The final reaction is catalyzed by thymidine phosphorylase (TP), which is expressed to a greater extent in some tumors than in normal tissues [18]. Therefore, cytotoxic 5-FU is generated to a higher degree in tumor cells than off-target tissues, which makes capecitabine a low-toxicity chemotherapeutic drug [18]. TP expression levels are diverse in different tumor types [18]; this limits the use of capecitabine to only several types of cancers. In present study, we assessed the anticancer effect of DCA in combination with capecitabine for cancers that modestly expressed TP. We hypothesized that DCA increases the anticancer effects and reduces the effective dose of capecitabine. The combination of DCA with capecitabine might produce a good treatment regimen because both agents can be taken orally with good patient adherence. Furthermore, generic forms of DCA may reduce the effective dose of capecitabine, thus decreasing side effect and the cost of cancer treatment.
Materials and methods
Materials
Sodium dichloroacetate (DCA, CSA:2156-56-1), purity 99 %, was obtained from Shanghai Jieshi Chemical Co., Ltd. (China). 5-Fluorouracil (5-FU) and 5′-deoxy-fluorouridine (5-DFUR) was obtained from Sigma-Aldrich (USA). MTT was obtained from Shanghai Biological Engineering Co., Ltd. (China). Capecitabine tablets (Xeloda) were obtained from Roche (USA). Mouse B16 melanoma and human non-small cell lung cancer A549 cell line were obtained from American Type Cell Culture Collection (ATCC, USA).
Animal model studies
Allograft model
C57BL/6 mice, female, 6–8 weeks old and weighing approximately 18–20 g, were purchased from Shanghai Laboratory Animal Center (SLAC, China) and allowed to acclimate for 1 week. One million B16 single cells were subcutaneously (s.c.) inoculated into the right flank of the C57BL/6 mice. Mice were randomly grouped, with 6 mice per group in a cage. There were two sets of mice. DCA and capecitabine were administered to these two sets of mice at 3 and 10 days after inoculation, respectively. DCA was added into sterile drinking water to a final concentration of 1.4 g/L. Measurement of the volume of water consumed showed that the amount of DCA administered to each mouse was approximately equal to 100 mg/kg/day. Capecitabine tablets were ground and suspended in sterile water with 4 % carboxymethyl cellulose to make different concentrations. Two hundred microliters of the capecitabine suspensions was intragastrically administered to each mouse. Every 2 days, the long (a) and short (b) diameters of the tumors were measured using a vernier caliper, and body weight was recorded. Tumor volume was calculated by the formula V = 0.5ab2. Twenty-two days after inoculation, the mice were killed and tumors were removed and weighed.
Xenograft model
BALB/c-nu mice, male, 5–6 weeks old and weighing approximately 18–20 g, were purchased from Shanghai Laboratory Animal Center (SLAC, China) and allowed to acclimate for 1 week. Approximately 2 × 2 mm sections of newly minced A549 tumor tissues, which were from BALB/c-nu mice previously inoculated with A549 cells, were inoculated s.c. into the right flank region of male BALB/c-nu mice. DCA and capecitabine were administered to mice when their tumor volume reached ~0.2 cm3. Thirty to thirty-five days after treatment, the mice were killed and tumors were removed and weighed. Other methods were the same as those described in the allograft experiment.
The animal studies were approved by Animal Welfare and Ethics Group of Laboratory Animal Science Department, Fudan University.
Histology and immunohistochemistry
Histological examination of the tumor nodules was performed using additional animals (3 mice each group) that were not considered for the monitoring of tumor growth. Tissues were fixed in 4 % (w/v) paraformaldehyde, and after fixation over night at room temperature, samples were dehydrated in graded ethanol and embedded in paraffin. Thereafter, different parts of tumor were randomly cut to make 4-μm sections on the Leica microtome. After deparaffinization and rehydration, three sections from different parts of each sample were chosen for the follow-up operations. Terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) and 4′6-diamidino-2-phenylindole (DAPI) staining were performed according to manufacturer’s instructions of TUNEL and DAPI kits (Beyotime, China). Sections were analyzed using an inverted fluorescence microscope (Olympus, Japan). Detection of proliferating cell nuclear antigen (PCNA) was carried out after section deparaffinization and incubation at 96–100 °C for 20 min. Endogenous peroxidase activity was quenched with 0.3 % (v/v) hydrogen peroxide in 60 % (v/v) methanol for 30 min. Non-specific adsorption was minimized by incubating the sections in 2 % (v/v) normal goat serum in PBS for 20 min. Tissue sections were incubated overnight with rabbit polyclonal anti-PCNA antibody (Abcam; 1:100 in PBS), washed with PBS 3 times, 30 min per time, and incubated with a biotin-conjugated goat antirabbit IgG for 2 h at 37 °C and avidin–biotin–peroxidase complex for 1 h at 37 °C. Sections were counterstained with hematoxylin (Sigma-Aldrich) and analyzed by light microscopy (Olympus, Japan). Three tumors per group were used to immunohistochemistry analysis. One or two sections per tumor were blindly selected to measure PCNA- or TUNEL-positive cells. Five random fields per slide in 400× magnification were measured blindly (n = 250 cells per group). When carried out the quantitative analysis of TUNEL-positive cells, the possible necrotic cells were excluded out by observing the nuclear morphology with DAPI staining. Both positive and negative controls were performed to ensure accurate results.
Protein extraction and Western blot
Tumor tissues from each group (3 additional mice each group) were pooled together and ground under liquid nitrogen and then lysed in 150 μL of tissue lysis buffer (Beyotime, China). Tubes were shaken vigorously for 1 min, placed on ice for 20 min, and centrifuged at 5,000g for 5 min at 4 °C. Total protein concentration was determined using a BCA protein determination kit (BioRad). Thirty micrograms of total protein from each sample was separated by SDS-PAGE on a 10 % gel, transferred to PVDF membrane, blocked, incubated overnight with primary antibody, incubated for 1 h with secondary antibody, and developed with the chromogenic substrate NBT/BCIP. Mouse monoclonal anticaspase 3 antibody (1:500), anticaspase 9 antibody (1:1,000), anti-β-actin antibody (1:2,000), and rabbit polyclonal anticaspase 8 antibody (1:1,000) were obtained from Beyotime (Nanjing, China). Mouse monoclonal anti-TP antibody (1:2,000) was from Abcam (UK). β-Actin was used as internal control. Band density of Western blot was analyzed with Clinx Gel Analysis V2.02 (Clinx Science, China).
Real-time PCR
B16 tumors and liver tissues were resected from the same C57BL/6 mouse previously inoculated with B16 cells (3 mice were used, gift from Wenlong Ren at Shanghai Institute of Pharmaceutical Industry). Colo205/A549 and liver tissues were resected from the same BALB/c-nu mouse previously inoculated with Colo205/A549 cells (3 mice were used, respectively, gift from Wenlong Ren at Shanghai Institute of Pharmaceutical Industry). For analysis of TP expression after treatment, tumor samples were pooled together from 3 equal weight tumor tissues each group. Total RNA was isolated with Trizol reagent (Invitrogen, USA) and reverse transcribed with PrimeScript® RT reagent kit (Takara, Japan). cDNA was normalized with β-actin. Real-time PCR was performed by three-step methods using SYBR® Premix Ex Taq™ II kit (Takata, Japan) with 55 °C annealing temperature and 40 amplification cycles. Individual test was carried out in triplicate. β-actin was used as internal control. The relative amount of each cDNA was analyzed by means of 2−△△Ct. Primers for real-time PCR: β-actin F: 5′-TCAAGATCATTGCTC CTCCTG-3′ and β-actin R: 5′-CTGCTTGCTGATCCACATCTG-3′, hTP F: 5′-TGGCTCAGTCGGGACAGCAG-3′ and hTP R: 5′-TCCGCTGATCATTG GCACCT-3′, mTP F: 5′-GCCTAGCTAAAGCATTGTGCTC-3′ and mTP R: 5′-AAGGGTGCT CGATCTGATAGCA-3′.
Statistics
We used multiple comparisons ANOVA with post hoc analysis (Tukey’s test), using SPSS 16.0 software (SPSS Inc., USA). Data are presented as mean ± SEM, and P < 0.05 was considered significant.
Results
TP expression in mouse B16 melanoma and human A549 NSCLC tumors
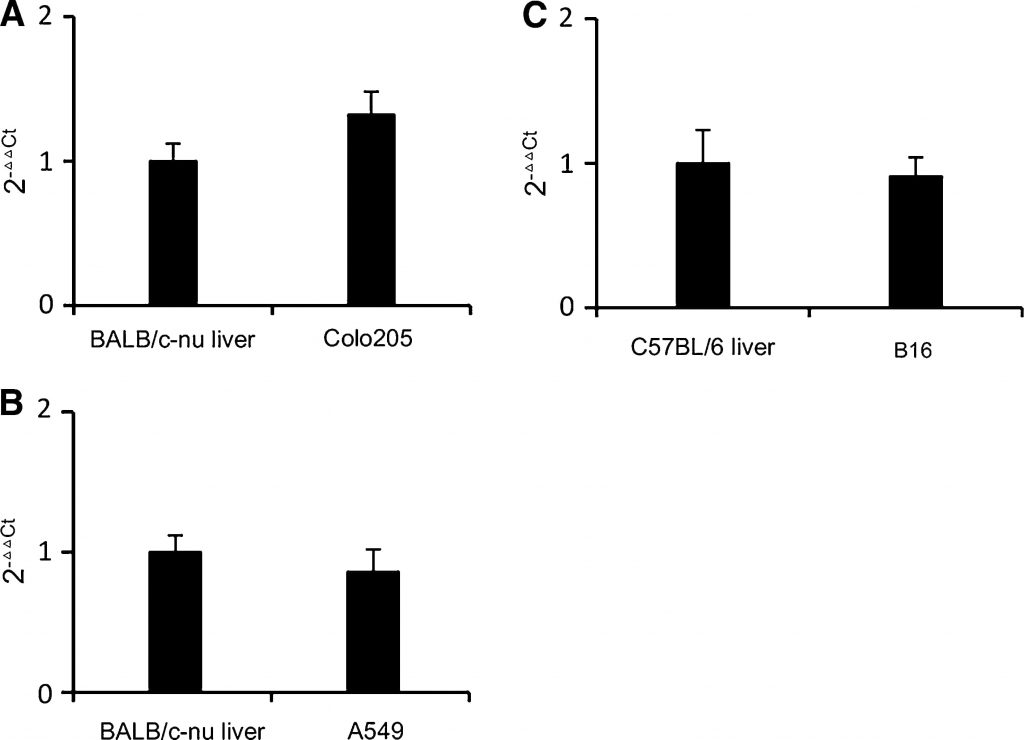
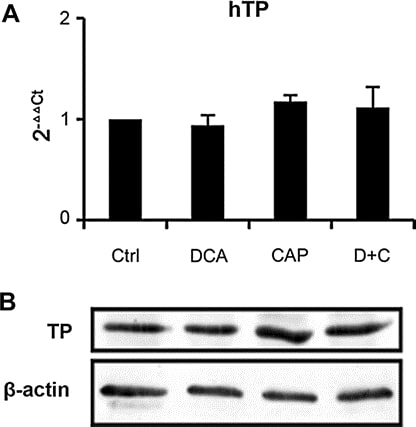
TP expression in B16 and A549 tumors removed from mice was analyzed by real-time PCR. Human livers express relatively more TP than other normal tissues [18]. In a typical cancer type that suitable for capecitabine treatment, TP expression in tumor is close to or higher than livers, colorectal and breast cancers for example [18,19]. In human cancer xenografts, cancers with high TP activity were more susceptible to capecitabine treatment than those with low TP activity [20]. Colorectal cancer cell line Colo205 was chosen as reference based on its moderate TP expression and moderate sensitivity to capecitabine [21]. As shown in Fig. 1a, TP transcription levels in Colo205 were a little higher than in liver of BALB/c-nu mouse. As shown in Fig. 1b, c, TP transcription levels in B16 and A549 tumors were close to those in the mouse livers. Therefore, B16 and A549 were also moderately TP-expressing cell lines. We can deduce that B16 and A549 will respond to capecitabine treatment to some extent without covering the effect of DCA. So, B16 allograft and A549 xenograft models were suitable to study the antitumor effect of DCA and capecitabine in combination.
DCA increases the antitumor effect of capecitabine in a mouse melanoma B16 allograft without additional toxicity
As the hypoxic nature of the tumor microenvironment is critical for optimal DCA activity, the antitumor effect of DCA plus capecitabine was tested in animal models instead of cell lines. Thirty-six C57BL/6 mice were inoculated with 1 × 106 B16 melanoma cells and randomly separated into 6 groups (n = 6): a control group that received no drug, a DCA alone group, a 10 mg/day capecitabine alone group, and 3 groups of DCA plus either 5, 10, or 20 mg/day capecitabine. Three days after tumor cell inoculation, DCA in drinking water and oral (p.o.) capecitabine as single agents or in combination with escalating capecitabine concentrations were administered to mice. As shown in the upper panel of Fig. 2a, both DCA and 10 mg/day capecitabine alone treatments modestly inhibited the growth of B16 melanoma tumors compared to control group. In contrast, DCA plus 10 mg/day capecitabine significantly increased tumor growth inhibition (P < 0.05). The antitumor effect of DCA plus 20 mg/day capecitabine was similar to that seen with DCA plus 10 mg/day capecitabine; tumor growth was almost completely inhibited. It is noteworthy that mice treated with DCA plus 20 mg/day capecitabine experienced an acute lose of body weight (Fig. 2a, lower panel), while DCA plus 10 mg/day capecitabine had little effect on body weight when compared to control.
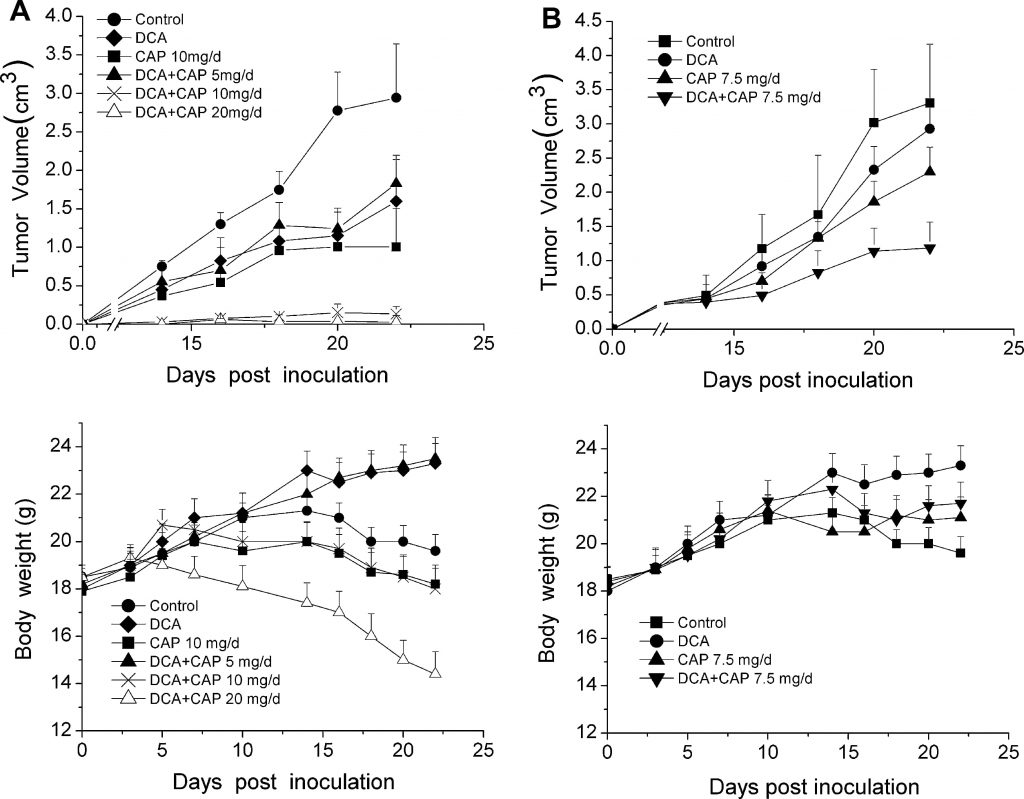
To assess the antitumor effect of DCA plus capecitabine against palpable, detectable tumors, 10 days after tumor cell inoculation, DCA and capecitabine alone or in combination were administered to a second group of 36 C57BL/6 mice inoculated with 1 × 106 B16 melanoma cells. Mice with palpable tumors were randomly separated into 4 groups (n = 9, 3 mice were used for immunohistochemistry analysis), control, DCA alone, capecitabine alone at 7.5 mg/day, and DCA plus capecitabine at 7.5 mg/day. As shown in upper panel of Fig. 2b, DCA plus 7.5 mg/day capecitabine significantly inhibited tumor growth compared to either DCA or capecitabine alone (P < 0.05). Twenty-two days after inoculation, DCA plus 7.5 mg/day capecitabine inhibited tumor growth by 75 % (P < 0.05), while DCA and capecitabine alone only inhibited growth by 25 % and 35 %, respectively (P < 0.05). DCA did not cause any acute loss in body weight compared to capecitabine treatment alone (Fig. 2b, lower panel). These results show that DCA and capecitabine may have a synergetic antitumor effect in B16 melanoma tumors.
DCA increases the antitumor effect of capecitabine in a human NSCLC A549 xenograft model without additional toxicity
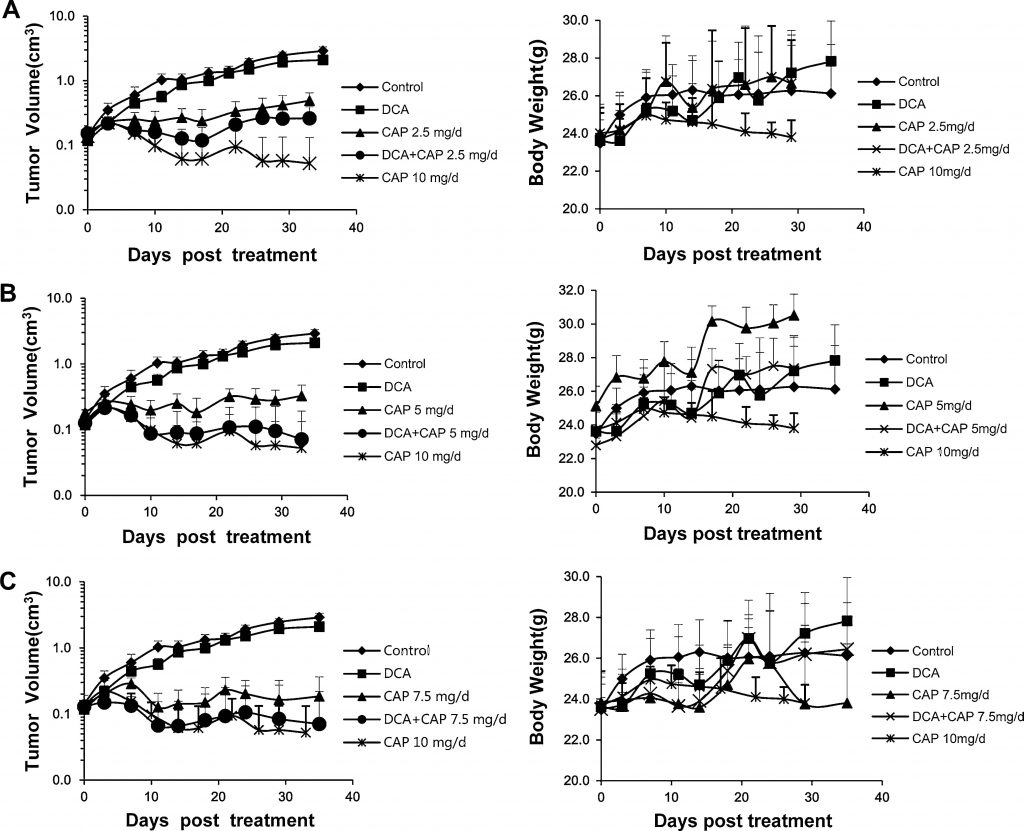
It has been previously reported that human NSCLC could be treated with either DCA [7] or capecitabine [22–24]. In the present study, we investigated the antitumor effect of DCA plus capecitabine in a human NSCLC A549 xenograft model. Sixty-six male BALB/c-nu mice with human NSCLC A549 tumors (~2 × 2 mm) inoculated s.c. into the right flank were randomly separated into 9 groups: control; DCA alone; 2.5, 5, 7.5, or 10 mg/day capecitabine alone; or DCA plus 2.5, 5, or 7.5 mg/day capecitabine (n = 6, except control, DCA alone, 7.5 mg/day capecitabine and DCA plus 7.5 mg/day capecitabine; in these groups, n = 9, 3 mice were used for immunohistochemistry analysis and Western blot or real-time PCR analysis). Capecitabine at 10 mg/day was set as a high-dose control. When tumor volumes reached 0.15–0.2 cm3, drugs were administered to mice. Capecitabine was administered p.o. in a 14-day on/7-day off schedule. As shown in the left panels of Fig. 3a, b, and c, DCA alone had a slight growth-inhibiting effect on A549 tumors; this finding was inconsistent with those of previous reports [7] in which DCA alone showed a greater antitumor effect. Capecitabine alone at 10 mg/day robustly inhibited the growth of A549 tumors, but a dramatic loss in body weight was noticed, implying severe toxicity (Fig. 3a, b, c, right panels). As shown in the left panel of Fig. 3a, 2.5 mg/day of capecitabine alone significantly reduced the growth of A549 tumors; the combination of DCA plus 2.5 mg/day capecitabine increased the growth inhibition. The rising tumor volume curve of DCA alone treatment suggests that capecitabine exerts the dominant antitumor effect in combination treatment. The effect of DCA plus 5 mg/day capecitabine was slightly better than DCA plus 2.5 mg/day capecitabine, although worse than 10 mg/day capecitabine alone, and no significant loss in body weight was observed (Fig. 3b, right panel). It is noteworthy that DCA plus 5 mg/day capecitabine significantly increased tumor growth inhibition compared to 5 mg/day capecitabine alone (Fig. 3b, left panel). The antitumor effect of the combination was close to the 10 mg/day capecitabine treatment, but without significant loss in body weight (Fig. 3b, right panel). The effect of 7.5 mg/day capecitabine alone (Fig. 3c) was slightly better than 5 mg/day capecitabine alone; however, when combined with DCA, there was no significant difference between DCA plus 7.5 mg/day capecitabine and DCA plus 5 mg/day capecitabine. These results imply that DCA can reduce the dose of capecitabine without losing the antitumor effects or increasing toxicity.
DCA increases the apoptotic effect of capecitabine on B16 and A549 cells in vivo
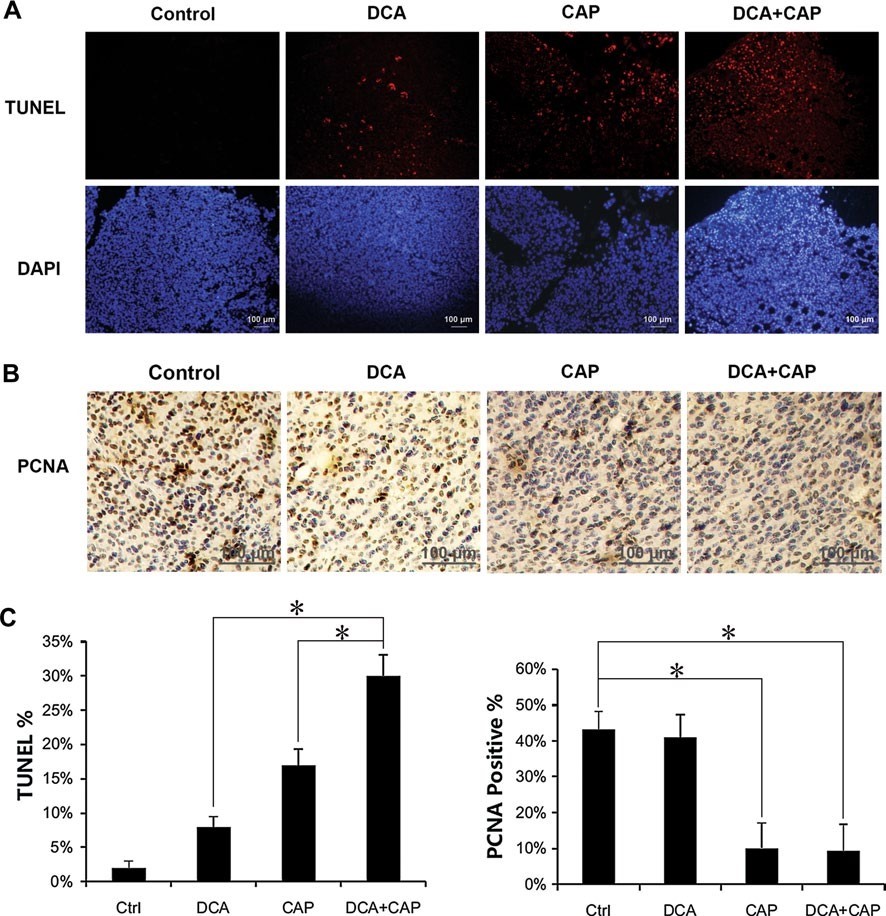
Histological examination of the B16 melanoma tumors was performed 7 days after the beginning of treatment. TUNEL staining revealed that treatment for B16 melanoma tumors with either DCA or 7.5 mg/day capecitabine alone induced cellular apoptosis to 8 and 17 %, respectively. In combination, DCA plus 7.5 mg/day capecitabine induced apoptosis to approximately 30 %, more than the sum of the single treatments combined (Fig. 4a, c, left panel). Staining of PCNA in the B16 melanoma tumors revealed that DCA alone had little impact on proliferation, while 7.5 mg/day capecitabine alone significantly reduced the proliferation. DCA plus 7.5 mg/day capecitabine did not increase the proliferation inhibition of capecitabine alone in the B16 tumor cells (Fig. 4b, c, right panel).
Similar to the B16 melanoma tumors, treatment for NSCLC A549 tumors with DCA or 7.5 mg/day capecitabine alone induced apoptosis to 15 and 30 %, respectively. When combined together, DCA plus 7.5 mg/day capecitabine induced apoptosis to 50 %, more than the sum of the single agent treatments combined (Fig. 5a, b). These results suggest that DCA and capecitabine have a synergetic effect on NSCLC A549 tumor cell apoptosis. DCA did not increase the proliferation inhibition of capecitabine in NSCLC A549 tumor cells (data not shown).
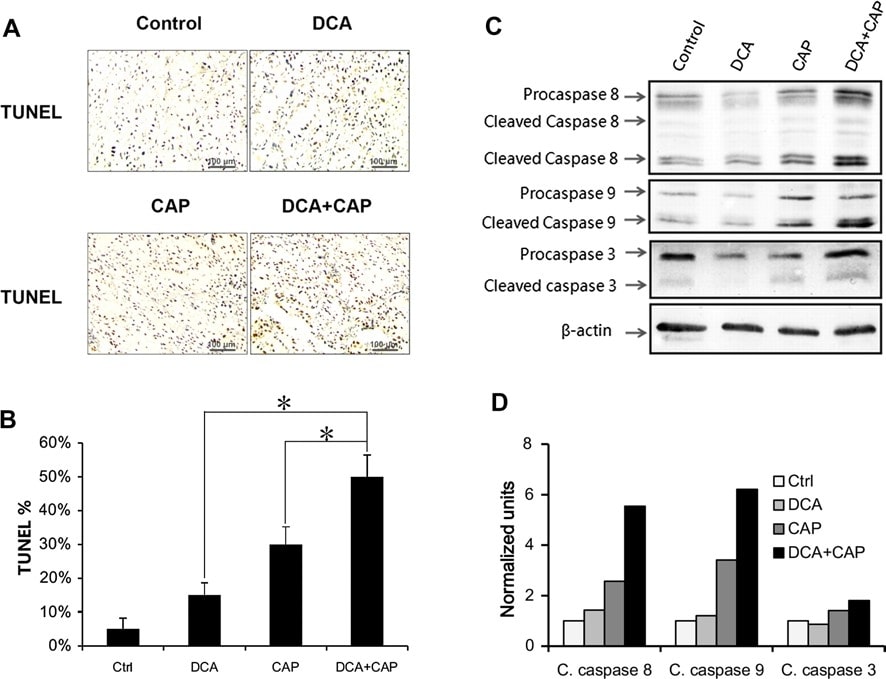
Western blot showed that DCA had little effect on the expression and activation of procaspase 8, procaspase 9, and procaspase 3 in NSCLC A549 tumors compared to control at day 7 after treatment (Fig. 5c, d). Capecitabine alone at 7.5 mg/day increased the activation of all 3 procaspases (Fig. 5c, d). Interestingly, although DCA alone had little effect on the expression and activation of the 3 procaspases, DCA plus capecitabine promoted the expression of procaspase 8 and procaspase 3 compared to capecitabine alone treatment and increased the activation of procaspase 8, procaspase 9, and procaspase 3.
DCA has little effect on the expression of TP in tumor
We analyzed the expression of TP in tumor samples from A549 xenograft at day 7 post-treatment. Both real-time PCR (Fig. 6a) and Western blot (Fig. 6b) showed that DCA as single agent or in combination with capecitabine has little effect on TP expression, which means the mechanism of DCA and capecitabine in combination was different from other capecitabine synergist previous reported [19,25–27]. Real-time PCR also showed that DCA did not affect the expression of other capecitabine metabolic enzymes (thymidylate synthase, orotate phosphoribosyl transferase, dihydropyrimidine dehydrogenase, thymidine kinase 1, and cytidine deaminase, data not shown). The results suggest that DCA has little effect on the metabolism of capecitabine, which would not increase toxicity of capecitabine.
Discussion
DCA alone or in combination with other therapies has been tested in clinical trials; however, no reports on the antitumor effects of DCA in combination with capecitabine are available. We hypothesized that the apoptosis-promoting effect of DCA on solid tumor cells would make them more sensitive to capecitabine. In the present study, 1.4 g/L DCA alone had little effect on NSCLC A549 tumor growth in mice. However, combined administration of DCA and capecitabine was able to reduce the effective dose of capecitabine by 50 %. DCA increased the antitumor effect of capecitabine in vivo via sensitizing apoptosis. DCA has little effect on the metabolism of capecitabine, which would not increase toxicity of capecitabine.
As both positive and negative results have been reported (as shown in Introduction section), there is still controversy over the use of DCA as an anticancer agent. This discrepancy may be due to different experimental conditions, particularly between in vivo and in vitro results. The cellular assays used in present study showed that tumor cells are insensitive to DCA when cultured in vitro, IC50 is above 50 mM. (data not shown). The effect of DCA on cancer cells is not due to direct cytotoxity but depends on the metabolic pattern of cancer cells [7, 28]. Cell lines cultured in vitro cannot replicate the tumor microenvironment and may lose the “Warburg effect”. Therefore, cellular-based in vitro assays may be not the most relevant model systems to determine the use of DCA. That is why the combined antitumor effect of DCA and capecitabine was evaluated in mouse tumor models in vivo firstly in present study. We also found that the effect of DCA on the NSCLC A549 xenograft in present study was less than that reported by Bonnet et al. [7], even though a larger dose of DCA was used. This may be due to the difference in the metabolic capacity of DCA between mice and rats. The results suggest that the effect of DCA may be affected by the state of cancer cells and patients, which calls for careful study in clinical use of DCA in cancer treatment.
In present study, animal model studies demonstrated that DCA itself showed mild antitumor effect against established tumors. But when combined with capecitabine, DCA dramatically increased the antitumor effect of capecitabine, which can be seen in both animal model studies and apoptosis immunohistochemistry results. Consistent with that, Western blot analysis showed that DCA itself had little effect on the expression and cleavage of procaspases. But when combined with capecitabine, DCA remarkably increases the expression and cleavage of procaspases 8, 9, and 3. It is reported that cytotoxic agent, like 5-Fu, can lead to increased expression and activation of caspase 8 [29,30]. The reason why capecitabine leads to increased expression and activation of caspase 9 is still not clear. It might be related to the antiangiogenetic effect of capecitabine. It is reported that DCA can normalize a mitochondria-K+ channel axis and act as an apoptosis sensitizer [7]. We speculate that DCA may depolarize mitochondrial potential of cancer cells by normalizing the axis, therefore augment the activation of caspase 8 and caspase 9 by capecitabine. Increased activation of caspase 8 and caspase 9 leads to increased activation of caspase 3.
Recently, the critical contribution of CSCs, with their enhanced tumorigenic ability and resistance to radiotherapy and chemotherapy, to malignant behavior has been highlighted [31]. CSCs in solid tumors are reported to play an important role in the antichemotherapy and antiradiotherapy characteristics of tumors [32, 33]. Many therapies to target CSCs have been discussed [34, 35], and combination therapies to target both CSCs and “normal” cancer cells have aroused great interest [36–38]. It is reported that DCA induced apoptosis in putative glioblastoma stem cells, both in vitro and in vivo [9]. In our study, after 7 days of DCA treatment, the number of CD133-positive cells in the A549 tumor slides determined by immunohistochemistry reduced to 0.5 % compared to 6 % in control group (data not shown), which leads us to speculate that DCA may also have effect on inducing apoptosis in lung cancer CSCs. We speculate that DCA may sensitize tumor cells, especially CSCs, for capecitabine. The combination of DCA and capecitabine works in 2 ways against tumor cells; capecitabine targets the “normal” cancer cells, sparing the CSCs, and DCA targets the CSCs and promotes apoptosis to capecitabine. Another likely scenario to explain the combination effects is that DCA suppressed angiogenesis of cancer in vivo [9], in which DCA does not show direct effect on cancer cells. In addition to its classic antitumor activity, capecitabine may also act as an antiangiogenetic molecular according to recent studies [39]. DCA may augment the antiangiogenic effect of capecitabine, which explains the combination antitumor effect. In-depth studies will be done to determine the detailed mechanism.
In conclusion, we used both syngenetic-grafted tumor and xenografted tumor models to study the combination antitumor effect of DCA and capecitabine and found that DCA potentiated the antitumor effect of capecitabine for the first time. We determined that DCA had the ability to sensitize cancer cells and increased the apoptotic effects of capecitabine. When given in combination, DCA was able to reduce the effective dose of capecitabine without increased toxicity. The low-cost, generic, and oral DCA in combination with oral capecitabine may be a good therapy regimen against cancers.
Acknowledgments
We are very grateful to Wenlong Ren at Shanghai Institute of Pharmaceutical Industry for the assistance in the preparation of the mouse tumor models.
Conflict of interest
None.
REFERENCES
1 Wigfield SM, Winter SC, Giatromanolaki A, Taylor J, Koukourakis ML, Harris AL (2008) PDK-1 regulates lactate production in hypoxia and is associated with poor prognosis in head and neck squamous cancer. Br J Cancer 98(12):1975–1984. doi:10.1038/sj.bjc.6604356
2 Cohen RD, Iles RA (1978) Dichloroacetate and the treatment of lactic acidosis. New Engl J Med 298(24):1364. doi:10.1056/NEJM197806152982413
3 Agbenyega T, Planche T, Bedu-Addo G, Ansong D, Owusu-Ofori A, Bhattaram VA, Nagaraja NV, Shroads AL, Henderson GN, Hutson AD, Derendorf H, Krishna S, Stacpoole PW (2003) Population kinetics, efficacy, and safety of dichloroacetate for lactic acidosis due to severe malaria in children. J Clin Pharmacol 43(4):386–396
4 Michelakis ED, Webster L, Mackey JR (2008) Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer 99(7):989–994. doi:10.1038/sj.bjc.6604554
5 Warburg O, Wind F, Negelein E (1927) The metabolism of tumors in the body. J General Physiol 8(6):519–530
6 Gatenby RA, Gillies RJ (2004) Why do cancers have high aerobic glycolysis? Nat Rev Cancer 4(11):891–899. doi:10.1038/nrc1478
7 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED (2007) A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11(1):37–51. doi:10.1016/j.ccr.2006.10.020
8 Pan JG, Mak TW (2007) Metabolic targeting as an anticancer strategy: dawn of a new era? Science’s STKE: signal transduction knowledge environment 2007(381):pe14. doi:10.1126/stke.3812007pe14
9 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, Petruk KC (2010) Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2(31):31ra34. doi:10.1126/scitranslmed.3000677
10 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ (2008) Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate 68(11):1223–1231. doi:10.1002/pros.20788
11 Xiao L, Li X, Niu N, Qian J, Xie G, Wang Y (2010) Dichloroacetate (DCA) enhances tumor cell death in combination with oncolytic adenovirus armed with MDA-7/IL-24. Mol Cell Biochem 340(1–2):31–40. doi:10.1007/s11010-010-0397-6
12 Kwitniewski M, Moan J, Juzeniene A (2011) Metabolic-targeted therapy with dichloroacetate (DCA): a novel treatment strategy to improve the outcome of photodynamic therapy. Photochem Photobiol Sci Off J Eur Photochem Assoc Eur Soc Photobiol 10(1):25–28. doi:10.1039/c0pp00193g
13 Tong J, Xie G, He J, Li J, Pan F, Liang H (2011) Synergistic antitumor effect of dichloroacetate in combination with 5-fluorouracil in colorectal cancer. J Biomed Biotechnol 2011:740564. doi:10.1155/2011/740564
14 Shahrzad S, Lacombe K, Adamcic U, Minhas K, Coomber BL (2010) Sodium dichloroacetate (DCA) reduces apoptosis in colorectal tumor hypoxia. Cancer Lett 297(1):75–83. doi:10.1016/j.canlet.2010.04.027
15 Heshe D, Hoogestraat S, Brauckmann C, Karst U, Boos J, Lanvers-Kaminsky C (2011) Dichloroacetate metabolically targeted therapy defeats cytotoxicity of standard anticancer drugs. Cancer Chemother Pharmacol 67(3):647–655. doi:10.1007/s00280-010-1361-6
16 Mandelblat J, Bashir T, Budman DR (2006) Capecitabine-docetaxel combination treatment. Expert Rev Anticancer Ther 6(9):1169–1178. doi:10.1586/14737140.6.9.1169
17 Budman DR (2000) Capecitabine. Invest New Drugs 18(4):355–363
18 Miwa M, Ura M, Nishida M, Sawada N, Ishikawa T, Mori K, Shimma N, Umeda I, Ishitsuka H (1998) Design of a novel oral fluoropyrimidine carbamate, capecitabine, which generates 5-fluorouracil selectively in tumours by enzymes concentrated in human liver and cancer tissue. Eur J Cancer 34(8):1274–1281
19 Endo M, Shinbori N, Fukase Y, Sawada N, Ishikawa T, Ishitsuka H, Tanaka Y (1999) Induction of thymidine phosphorylase expression and enhancement of efficacy of capecitabine or 5′-deoxy-5-fluorouridine by cyclophosphamide in mammary tumor models. Int J cancer J int du cancer 83(1):127–134
20 Ishikawa T, Sekiguchi F, Fukase Y, Sawada N, Ishitsuka H (1998) Positive correlation between the efficacy of capecitabine and doxifluridine and the ratio of thymidine phosphorylase to dihydropyrimidine dehydrogenase activities in tumors in human cancer xenografts. Cancer Res 58(4):685–690
21 Kolinsky K, Shen BQ, Zhang YE, Kohles J, Dugan U, Zioncheck TF, Heimbrook D, Packman K, Higgins B (2009) In vivo activity of novel capecitabine regimens alone and with bevacizumab and oxaliplatin in colorectal cancer xenograft models. Mol Cancer Ther 8(1):75–82. doi:10.1158/1535-7163.MCT-08-0596
22 Lee DH, Han JY, Yoon SM, Lee JJ, Lee HG, Kim HY, Yoon SJ, Hong EK, Lee JS (2006) A pilot trial of gemcitabine and vinorelbine plus capecitabine in locally advanced or metastatic nonsmall cell lung cancer. Am J Clin Oncol 29(2):143–147. doi:10.1097/01.coc.0000203743.32845.40
23Lee JJ, Han JY, Lee DH, Kim HY, Chun JH, Lee HG, Yoon SM, Lee SY, Lee JS (2006) A phase II trial of docetaxel plus capecitabine in patients with previously treated non-small cell lung cancer. Jpn J Clin Oncol 36(12):761–767. doi:10.1093/jjco/hyl106
24 Kindwall-Keller T, Otterson GA, Young D, Neki A, Criswell T, Nuovo G, Soong R, Diasio R, Villalona-Calero MA (2005) Phase II evaluation of docetaxel-modulated capecitabine in previously treated patients with non-small cell lung cancer. Clin Cancer Res off J Am Assoc Cancer Res 11(5):1870–1876. doi:10.1158/1078-0432.CCR-04-1727
25 Sawada N, Ishikawa T, Fukase Y, Nishida M, Yoshikubo T, Ishitsuka H (1998) Induction of thymidine phosphorylase activity and enhancement of capecitabine efficacy by taxol/taxotere in human cancer xenografts. Clin Cancer Res Off J Am Assoc Cancer Res 4(4):1013–1019
26 Sawada N, Kondoh K, Mori K (2007) Enhancement of capecitabine efficacy by oxaliplatin in human colorectal and gastric cancer xenografts. Oncol Rep 18(4):775–778
27 Sawada N, Ishikawa T, Sekiguchi F, Tanaka Y, Ishitsuka H (1999) X-ray irradiation induces thymidine phosphorylase and enhances the efficacy of capecitabine (Xeloda) in human cancer xenografts. Clin Cancer Res Off J Am Assoc Cancer Res 5(10):2948–2953
28 Kumar A, Kant S, Singh SM (2012) Novel molecular mechanisms of antitumor action of dichloroacetate against T cell lymphoma: implication of altered glucose metabolism, pH homeostasis and cell survival regulation. Chem Biol Interact 199(1):29–37. doi:10.1016/j.cbi.2012.06.005
29 Milner AE, Palmer DH, Hodgkin EA, Eliopoulos AG, Knox PG, Poole CJ, Kerr DJ, Young LS (2002) Induction of apoptosis by chemotherapeutic drugs: the role of FADD in activation of caspase-8 and synergy with death receptor ligands in ovarian carcinoma cells. Cell Death Differ 9(3):287–300. doi:10.1038/sj.cdd.4400945
30 Ehrhardt H, Hacker S, Wittmann S, Maurer M, Borkhardt A, Toloczko A, Debatin KM, Fulda S, Jeremias I (2008) Cytotoxic drug-induced, p53-mediated upregulation of caspase-8 in tumor cells. Oncogene 27(6):783–793. doi:10.1038/sj.onc.1210666
31 Ghotra VP, Puigvert JC, Danen EH (2009) The cancer stem cell microenvironment and anti-cancer therapy. Int J Radiat Biol 85(11):955–962. doi:10.3109/09553000903242164
32 Kitamura H, Okudela K, Yazawa T, Sato H, Shimoyamada H (2009) Cancer stem cell: implications in cancer biology and therapy with special reference to lung cancer. Lung Cancer 66(3):275–281. doi:10.1016/j.lungcan.2009.07.019
33 Dylla SJ, Beviglia L, Park IK, Chartier C, Raval J, Ngan L, Pickell K, Aguilar J, Lazetic S, Smith-Berdan S, Clarke MF, Hoey T, Lewicki J, Gurney AL (2008) Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PLoS One 3(6):e2428. doi:10.1371/journal.pone.0002428
34 Tao H, Zhu Y (2011) Colorectal cancer stem cell: a potential therapeutic target. Clin Transl Oncol Off Publ Fed Span Oncol Soc Natl Cancer Inst Mexico 13(12):833–838. doi:10.1007/s12094-011-0743-5
35 Tang C, Ang BT, Pervaiz S (2007) Cancer stem cell: target for anti-cancer therapy. FASEB J Off Publ Fed Am Soc Exp Biol 21(14):3777–3785. doi:10.1096/fj.07-8560rev
36 Sun XY, Nong J, Qin K, Warnock GL, Dai LJ (2011) Mesenchymal stem cell-mediated cancer therapy: a dual-targeted strategy of personalized medicine. World J Stem Cells 3(11):96–103. doi:10.4252/wjsc.v3.i11.96
37 Sapi E (2009) Novel cancer stem cell therapy on the horizon. Cancer Biol Ther 8(18):1754–1755
38 Shigdar S, Lin J, Li Y, Yang CJ, Wei M, Zhus Y, Liu H, Duan W (2012) Cancer stem cell targeting: the next generation of cancer therapy and molecular imaging. Ther Deliv 3(2):227–244
39Ranieri G, Roccaro AM, Vacca A, Ribatti D (2006) Thymidine phosphorylase (platelet-derived endothelial cell growth factor) as a target for capecitabine: from biology to the bedside. Recent Pat Anti-Cancer Drug Discov 1(2):171–183
Related content: